

Year: 2002 Vol. 68 Ed. 2 - (18º)
Artigo de Revisão
Pages: 268 to 275
 PDF PT
PDF PT Hearing loss related to mitochondrial DNA changes.
Author(s):
Maria F. P. de Carvalho 1,
Fernando A. Quintanilha Ribeiro 2.
Keywords: genetic disease, hearing loss, mitochondrial DNA
Abstract:
Hearing loss is a common symptom that may be manifested by many etiologies and it is frequently associated to genetic problems. Genetic mutations may occur in nuclear or mitochondrial genes. Mitochondria are intracellular organelles that have their own genome (DNA); mitochondrial DNA is from exclusive maternal inheritance. Although mitochondrial DNA mutations derive from maternal inheritance, spontaneous mutations may also occur. The phenotype is the clinical expression of a mitochondrial mutation and depends on the amount of mutant mitochondrial DNA contained in the cell. This phenomenon is known as heteroplasmy. The mitochondria provide energy to the cells by releasing ATP; thus, the greater the amount of energy required by the cell, the more likely it is to be affected by mitochondrial DNA mutations. Examples of high metabolism cells are nervous system cells, muscle cells, endocrine cells, optical and auditory cells. The cochlea has great energy turnover and mitochondrial DNA mutations of the hair cells will cause sensorineural hearing loss, which is normally bilateral, symmetrical and progressive. Hearing loss secondary to mitochondrial DNA mutations comprises 0.5 to 1% of all genetic hearing losses. Based on the literature review, it may be observed that hearing loss secondary to mitochondrial DNA mutations manifest in two distinct forms: isolated hearing loss (nonsyndromic), as in aminoglycoside hypersensitivity and presbyacusis, or associated to other diseases in a syndrome, such as Kearns-Sayre syndrome and maternally inherited diabetes and deafness.
![]()
Introduction
Hearing loss is a common symptom that may have different etiologies, among them, the genetic origin responsible for congenital and acquired hearing losses. Recent studied identified a series of mitochondrial DNA changes associated to hearing loss.
The purpose of the present study was to investigate the literature about syndromic and nonsyndromic cases related to some kind of mitochondrial DNA change.
a. Mitochondria
Mitochondria, cytoplasm organelles found in all mammal cells, have the primary role of transforming chemical energy of metabolites found in the cytoplasm into readily accessible energy to the cell. The energy is accumulated especially in components such as adenosine three-phosphate (ATP), which is used by the cell whenever it needs energy to produce osmotic, mechanical, electrical or chemical activities.
Mitochondria are spherical and elongated particles, 0.5 to 1 micron wide and 10 micra long. At electron microscopy there are two membranes - one smooth external one and another internal one with invaginations that form the mitochondrial crests. Each cell has 2 to 100 mitochondria that tend to gather in the cytoplasm where there is intense metabolic activity, such as the apical pole of hair cells.
b. Mitocondrial DNA
The mitochondria have their own DNA - mitochondrial DNA, which was discovered by Van Bruggen, Sinclair and Stevens & Nass, in 1966, but it was only completely sequenced in 1981, by Anderson et al1.
Mitochondrial DNA has a closed circular structure, with 16,569 paired bases and 37 genes. For oxidative phosphorylation, a process that takes place inside the mitochondria for the production of ATP, 74 polypeptides are needed and 13 are codified by the mitochondrial DNA - the other 61 are codified by nuclear DNA.
As to origin of mitochondrial DNA, there are two hypotheses: autogenous origin, in which both the mitochondria and the nucleus would originate from a process of compartmentalization and functional specialization inside the cell and the genome would originate from an already existing genome in the prokaryote cell. The exogenous origin, also known as the theory of endosymbiosis, believes that the mitochondria are originated from bacterial remains that had been phagocyted by the prokaryote cell. The remains would carry genetic material and the ability to metabolize the molecular oxygen present in the cytoplasm. At first, the bacteria were parasites, but as time went by and with evolution, the relations became symbiotic, forming the mitochondria. This is the most widely accepted theory, but it has not been confirmed yet.
Mitochondrial DNA has its own characteristics (Wallace, 19922): it is semi-autonomous, has independent genome replication, transcription and translation system; maternal heritage, mitochondrial DNA comes from the mother because the mitochondria present in the spermatozoa are located in the tail, which does not penetrate the egg during fecundation. Thus, mitochondria present in the embryo come exclusively from the mother. This characteristic of mitochondrial DNA has been used in anthropological studies and it reinforces the theory that the origin of mankind in Africa. The maternal heritage of mitochondrial DNA takes us to a female ancestor found in Africa, known as the sub-Saharan African Eva or mitochondrial Eva, who would be the female ancestor of us all, despite the fact that geneticists believe that it is a group of female ancestors, known as daughters of Eva, and not only one subject. Based on the mitochondrial DNA of African Eva, it was possible to study the migration lines of all populations in the world:
· Replicate segregation: when there is a mutation of mitochondrial DNA, the mutation may be present in the mitochondria of all body cells, a situation called homoplasmy, and upon cell division, the daughter cells will have mutant mitochondrial DNA. In another situation known as heteroplasmy, mutation may be present in only some mitochondria of a cell of a specific organ or tissue; and upon cellular division, the cell may segregate the mutant mitochondrial DNA and transmit to daughter cells little or no amount of mutant mitochondrial DNA. In such case, a mother may or may not transmit a mitochondrial mutation to her children.
· Expression threshold: the phenotype of a mitochondrial disease depends on the severity of the mutation and the need to produce energy in the involved organ.
· High rate of mutation: since mitochondrial DNA is small and compact, it may develop 10 to 20 times faster than nuclear DNA, increasing the likelihood of having a new pathogenic mutation.
There are four types of mutations that may occur at the mitochondrial DNA:
· Substitution of amino acids: rare condition, present in ophthalmologic and neurological diseases;
· Substitution of nucleotides: in such cases, the nucleotide of a specific locus is replaced by another one, for example, the adenine nucleotide is replaced by guanine or the cytosine nucleotide by thymine.
· Numeric alterations of deletion or duplication: deletion is the exclusion of a nucleotide from a specific locus, and the locus becomes empty. In duplication, the same locus is occupied by two nucleotides. These are sporadic mutations, not from maternal origin.
· Depletion: consists of global reduction of mitochondrial DNA genes. It is a rare fatal condition.
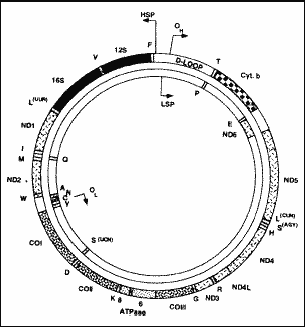
Figure 1. Structure of the circular molecule of DNAmt. OH (origin heavy) and OL (origin light) show the origin of replication of heavy and light chains, respectively. HSP and LSP correspond to transcription promoters of heavy and light chains; ND 1 to 6 are the genes that codify the sub-units of complex I of the respiratory chain; CO I to III are the genes that codify the sub-units of complex IV; ATPase 6 to 8 are the genes that codify the sub-units 6 to 8 of complex V; Cyt b is the gene that codifies cytochrome b of complex III; genes tRNA are indicated by one single letter that corresponds to amino acids codified by them; 12S and 16S are the genes that codify rRNA (Zeviani et al., 199829).
literature review
A. Hearing loss of mitochondrial origin
In genetic hearing losses, nuclear DNA mutations may occur in Mendelian genes and in sex-linked genes. According to the literature, there are approximately 100 nuclear genes involved in the symptoms of hearing loss, and only 30 of them have been mapped.
The mitochondrial disease was first described by Luft et al3, in 1962, when the authors described the case of a patient who had hypermetabolism of non-thyroid origin, whose muscle biopsy showed cellular paracrystalline inclusions in the mitochondria, at optic microscopy.
The correlation between mitochondrial disease and hearing loss was established by Petty et al4, in 1986, when they described a patient with mitochondrial myopathy and hearing loss. The incidence of hearing loss of mitochondrial origin seems to represent 0.5% to 1% of all genetic-based hearing losses.
Mitochondrial-based hearing loss may be of two types: syndromic, if hearing loss is related to a multiple clinical picture, usually severe, or nonsyndromic, if deafness is the only symptom.
B - Hearing loss associated to syndromic pictures
Many mitochondrial diseases present extremely exuberant and varied clinical pictures that are repeated in a number of patients, comprising a syndrome. Chart 1 sums up all syndromes that had sensorineural hearing loss (SNHL) as a symptom, their clinical characteristics, mutant gene, type of mutation and SNHL characteristics.
We are going to comment on two syndromic pictures that we consider the most interesting to Otorhinolaryngologists.
1. Diabetes and maternal heritage
In 1989, Lemkes et al5 described a German family in which maternal relatives presented diabetes mellitus and sensorineural hearing loss (SNHL) whose onset was at the age of 30 years.
In 1992, Van Den Ouweland6, studying other families with diabetes and maternal inherited deafness found a mitochondrial DNA mutation common to all patients, which consisted of the substitution of nucleotide adenine for guanine in locus 3243 of leukin codifying tRNA gene (tRNAleu gene).
Diabetes and maternal inherited deafness amounts to 1.5% of all cases of diabetes in Japan and Holland.
Clinical characteristics of diabetes and deafness are well defined:
· insulin-dependent diabetes: upon onset, diabetes may be not insulin-dependent but it normally progresses to insulin-dependent type because the mitochondrial change affects the secretion of insulin by the pancreas;
· the heritage is exclusively maternal;
· patients are thin;
· age of onset is before 40 years;
· type of hearing loss is sensorineural, initially in high frequencies, progressive and with recruitment, suggesting purely cochlear affection.
2. Kearns-Sayre Syndrome
The syndrome was described by Kearns & Sayre, in 1958, with clinical picture of external ophthalmoplegia, SNHL and varied grades of degenerative disease and myopia, such as: block of cardiac branch, cerebellar ataxia, paralysis of 7th and 8th cranial nerves, facial, neck and extremity muscle weakness and increased protein levels in the CSF.
Zeviani et al7, in 1989, studied patients with the syndrome and found deletions of various mitochondrial DNA genes. Poultron et al8, in the same year, found duplications of various mitochondrial DNA genes.
C. Hearing loss associated to nonsyndromic pictures
The so-called nonsyndromic cases have mitochondrial-based SNHL as the only symptom present, normally bilateral and progressive.
We are going to describe only two of the most interesting cases to Otorhinolaryngologists.
1. Hypersensitivity to aminoglycosides
Aminoglycoside antibiotics were widely used in the 60's for the treatment of respiratory tract infections and some patients, after using small amounts of the antibiotics, already manifested hearing loss. For this reason, ototoxic effects of aminoglycoside antibiotics became known and their use was restricted in the 70's.
In 1989, Higashi9, who studied families with hearing loss after using aminoglycoside antibiotics, observed a pattern of maternal inheritance and suggested that some kind of mitochondrial DNA mutation could be related to the hypersensitivity to the antibiotic.
Hutchin et al10, in 1993, discovered mitochondrial DNA mutation related to hypersensitivity, which is the exchange of the nucleotide adenine by guanine in locus 1555 (A1555G) of gene 12S ribosome RNA (12S rRNA). This type of mutation is homoplasmic.
The hearing loss is sensorineural, bilateral and profound.
Not all cases of hypersensitivity to aminoglycoside are correlated with mitochondrial DNA mutation. According to Einsink et al11 (1998), the mutation of A1555G of gene 12S rRNA was found in only 17% of the North-American population with non-family based SNHL after normal doses of aminoglycoside use, showing that it is a rare mutation.
2. Presbyacusis
Seidman et al12 (1996) studied mitochondrial DNA extracted from the cochlea of six human temporal bones coming from the temporal bone laboratory of the University of Chicago. They knew that three cochleae were from presbyacusis patients and the other three were from patients with normal hearing. Mitochondrial DNA was extracted and amplified by the PCR technique. As a result, they found mitochondrial DNA deletion in locus 4977 of cytochrome b, in two of the three cochleae of patients with presbyacusis. The cochleae of normal hearing patients did not present the deletion.
According to the authors, high frequency sound hearing is a process that demands more energy than low frequency sound hearing. To that end, vascular stria is rich in mitochondria, especially on the basal spiral of the cochlea, explaining why there are more cases of high frequency SNHL in mitochondrial abnormalities.
The pathophysiology of this kind of alteration, according to the authors, consists of the fact that deletions of mitochondrial DNA are linked to oxidative damage. Oxygen free radicals (OFR) are produced in vivo during cellular breathing and work as a self-oxidation route for chemical and biological molecules. OFR are biological contaminants that may be induced by ionization and ultra-violet radiation. OFR inhibit DNA replication and transcription, reacting with lipids, proteins and nucleic acids; they are naturally formed as a result of the reactions of oxidative phosphorylation and they could significantly contribute to DNA damage in the aging process.Ver Quadro
Chart 1. Syndromes caused by DNAmt changes that present hearing loss as a clinical characteristic (summary of the literature).
Chart 2. DNAmt changes causing nonsyndromic SNHL (summary of the literature).
Discussion
Physiopathogenesis
Since mitochondria are organelles responsible for cellular breathing, mutations in their DNA do not cause malformations but rather functional changes in the affected cells.
Inner ear affections are not completely unknown: in all cases in which hearing loss was investigated the type of hearing loss was sensorineural and many authors suggested cochlear involvement13, 14, 15, 16, 17 18. Other authors went further and suggested compromise of outer hair cells8, 9, others referred to retrocochlear changes, more specifically in the cochlear nucleus of the brainstem20.
In fact, the physiopathogenesis is unknown and many theories have been suggested. Yamasoba et al18 (1996), who studied diabetes and maternal inherited deafness presented the following theory:
Mitochondrial DNA change, abnormality in mitochondrial protein synthesis, changes in oxidative phosphorylation, reduced formation of ATP, changes in ionic pumps, alterations in potassium, calcium and sodium balance, cell death.
It is important to point out that the authors mentioned the likelihood of having a progressive reduction of oxidative phosphorylation, not manifested clinically while there is enough production of ATP for good cellular functioning. As of the moment that there is significant reduction of ATP clinical symptoms are manifested and that, in our opinion, explains the late onset age and the progression of both diabetes and sensorineural hearing loss.
Histopathogenesis
The only well-known anatomic pathologic substrate of a mitochondrial disease is mitochondrial myopathy, which has the so-called ragged-red fibers whose name refers to the appearance of the fibers with granular degeneration after they are stained with modified Gomori trichrome. The red color represents proliferation of mitochondrial elements and the presence of fibers suggest impairment of the electron transport system of the respiratory chain in the mitochondria4, 21, 22. The association of mitochondrial myopathy and hearing loss is frequent, because the tissues that finalize mitosis during embryogenesis, known as pre-mitotic, such as nervous, muscular and auditory tissues, tend to present high levels of heteroplasmy owing to replicate segregation, exceeding the expression threshold and manifested by myopathy, encephalopathy and hearing loss19, 23.
Unfortunately, up to today, we found only two studies published about histopathogenesis of mitochondrial-based hearing loss. The first study is by Lindsay & Hinojosa24, published in 1976, and it described the characteristics of outer, middle and inner ears of a patient with Kearns-Sayre syndrome, in whom it was possible to study the temporal bone after her death, at the age of 19 years. In the temporal bone study, the authors observed that both cochleae presented advanced degeneration of Corti's organ, tectorial membrane and vascular stria, collapsed Reissner membrane, bone spiral lamina without nervous fibers, reduced vascular supply for membranous structures and reduction of 60 to 70% of spiral ganglion in all cochlear turns. After 23 years, Yamasoba et al18 published a similar study of a patient with diabetes and maternally inherited hearing loss - she had mutation A3243G, the most common one for patients with diabetes and deafness. Findings were quite similar, with degeneration of vascular stria and outer hair cells.
Mutation investigation
In case of suspicion of mitochondrial mutation, the mutation investigation can be performed with samples of leukocytes or muscle cells submitted to two techniques: PCR or polymerase chain reaction which is very much used to investigate nucleotide substitution, or the Southern blot technique, used especially to detect deletions.
Generally speaking, deletions are sporadic and substitutions of nucleotides are of maternal inheritance.
In the suspicion of a mutation of mitochondrial DNA of nucleotide substitution, we should always investigate lysine codifying tRNA (tRNAlys), leukin codifying tRNA (tRNAleu), serine codifying tRNA (tRNAser(ucn)) and gene RNA ribosome 12S (12S rRNA). These genes are important because in some cases the same mutations may cause completely different phenotypes. For example, in the case of tRNAleu the substitution of the nucleotide adenine by guanine in locus 3243 may cause three different phenotypes: chronic progressive external ophthalmoplegia, diabetes and maternally-inherited deafness and the syndrome known as MELAS, mitochondrial myopathy associated with encephalopathy, lactic acidosis and cerebral vascular accidents.
It is not known why the same mutation would cause such different phenotypes. One possible cause would be the degree of heteroplasmy: the larger the amount of mitochondrial DNA present in the cells, the more exuberant and severe the picture, and the greater the amount of mutant DNA, the milder the clinical picture. Another possibility is that mitochondrial DNA would be restricted to auditory and pancreatic cells, in the case of diabetes and deafness, and restricted to the ophthalmologic cells in the case of chronic progressive external ophthalmoplegia 6. According to Hammans et al25 (1995), nuclear DNA may influence the phenotypic expression of the mutation, modulating its effects.
Therefore, we can observe that there are genes of mitochondrial DNA whose mutations generate clinical pictures in which sensorineural hearing loss is a frequent symptom. In patients with sensorineural hearing loss, genetic cause is rarely well investigated in Brazil. Thus, for diagnostic purposes, we suggest that when faced by a case of hearing loss suggestive of maternal inheritance, we should always try to identify laboratory and clinical characteristics to make it coincide with one of the syndromic pictures; if hearing loss is the only symptom, it may be a nonsyndromic affection. Even if there is no history of maternal transmission, mitochondrial etiology should not be discarded, because it may be caused by mitochondrial DNA deletions.
Treatment and perspectives
The treatment of mitochondrial diseases is still in an investigation phase: some authors referred to gene therapy or laboratory production of cofactors11, 26 as treatment approaches for the future.
Gene therapy consists of the transfer of selected genes into the host; this transfer uses a vector that may be a virus with tropism for the target organ.
Cofactors are polypeptides necessary for the oxidative phosphorylation and through biochemical tests we can find out which cofactor is deficient by the mitochondrial DNA mutation - and then replace it for a synthetic cofactor, in order to restore oxidative phosphorylation and ATP production.
Genetic counseling is essential in cases of hypersensitivity to aminoglycosides 13.
Cochlear implant is a support measure valid for cases of sensorineural hearing loss of mitochondrial origin27, 28, 29.
Conclusions
After a vast literature review, we concluded that:
1. Hearing loss cases associated with mitochondrial DNA changes may be syndromic or nonsyndromic.
2. The prevalence of mitochondrial-based sensorineural hearing loss is low, amounting to approximately 0.5 to 1% of all genetic-based hearing losses.
3. Mitochondrial DNA may be influenced by nuclear DNA.
4. The phenotype of mitochondrial mutations may vary according to the degree of heteroplasmy.
5. The mutations of mitochondrial DNA are of maternal inheritance, but they can also occur spontaneously.
6. Pathophysiology of mitochondrial DNA mutation-based hearing loss is related to lack of ATP in an organ that requires great supply, such as the cochlea.
7. Mitochondrial-based hearing loss is sensorineural, bilateral, progressive and symmetrical. The severity and onset of loss are variable.
References
1. Anderson S, Bankier AT, Barrell BG, Bruijn MHL, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJH, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature, 1981;290:457-65.
2. Wallace DC. Diseases of the mitochondrial DNA. Annu. Rev. Biochem., 1992;61:1175-212.
3. Luft R, Ikkos D, Palmieri G, Ernster L, Afzelius B. A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: a correlated clinical, biochemical, and morphological study. J. Clin. Invest., 1962;41:1776-1804.
4. Petty RKH, Harding AE, Morgan-Hughes JA. The clinical features of mitochondrial myopathy. Brain, 1986;109:915-38.
5. Lemkes HHPJ, Vijlder M, Struyvenberg P, Van Der Kamp JJP, Frolich M. Maternal inherited diabetes-deafness of the young (MIDDY): a new mitochondrial syndrome. Diabetologia, 1989;32:509 A.
6. Van Den Ouweland JMW, Lemkes HHPJ, Ruitenbeek W, Sandkuijl LA, De Vijlder MF, Struyvenberg PA A, Van De Kamp JJP, Maassen JA. Mutation in mitochondrial tRNAleu(uur) gene in a large pedigree with maternally transmitted type II diabetes and deafness. Nat. Genet., 1992;1:368-71.
7. Zeviani M, Servidei S, Gellera C, Bertini E, Dimauro S, Didonato S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the d-loop region. Nature, 1989;339:309-11.
8. Poultron J, Deadman ME, Gardiner RM. Duplications of mitochondrial DNA in mitochondrial myopathy. Lancet, 1989;4:2369.
9. Higashi K. Unique inheritance of streptomycin-induced deafness. Clin. Genet., 1989;35:433-36.
10. Hutchin T, Haworth I, Higashi K, Fischel-Ghodsian N, Stoneking M, Saha N, Arnos C, Cortopassi G. A molecular basis for human hypersensitivity to aminoglycoside antibiotics. Nucleic Acids Res., 1993;21:4174-9.
11. Ensink RJH, Camp GV, Cremers RJ. Mitochondrial inherited hearing loss. Clin. Otolaryngol., 1998;23:3-8.
12. Seidman MD, Bai U, Khan MJ, Murphy MP, Quirk WS, Castora FJ, Hinojosa R. Association of mitochondrial DNA deletions and cochlear pathology: a molecular biologic tool. Laryngoscope, 1996;106:777-83.
13. Braverman I, Jaber L, Levi H, Adelman C, Arons KS, Fischel-Ghodsian N, Shorat M, Elidan J. Audiovestibular findings in patients with deafness caused by a mitochondrial susceptibility mutation and precipitated by an inherited nuclear mutation or aminoglycosides. Arch. Otolaryngol. Head Neck Surg., 1996;122:1001-4.
14. Fischel-Ghodsian N, Prezant TR, Fournier P, Stewart IA, Maw M. Mitochondrial mutation associated with nonsyndromic deafness. Am. J. Otolaryngol., 1995;16:403-8.
15. Jaber L, Shohat M, Bu X, Fischel-Ghodsian N, Yang HY, Wang SJ, Rotter JI. Sensorineural deafness inherited as a tissue specific mitochondrial disorder. J. Med. Genet., 1992;29:8690.
16. Swift AC, Singh SD. Hearing impairment and the Kearns-Sayre syndrome. J. Laryngol. Otol., 1988;102:626-7.
17. Vialettes B, Paquis-Flucklinger V, Bendahan D. Clinical aspects of mitochondrial diabetes. Diabete Metab., 1997;23:526.
18. Yamasoba T, Yoshimoto O, Tsukuda K, Nakamura M, Kaga K. Auditory findings in patients with maternally inherited diabetes and deafness harboring a point mutation in the mitochondrial transfer RNAleu(uur) gene. Laryngoscope, 1996;106:49-53.
19. Oshima T, Ueda N, Ikeda K, Abe K, Takasaka T. Bilateral sensorineural hearing loss associated with the point mutation in mitochondrial genome. Laryngoscope, 1996;106:43-8.
20. Tiranti V, Chariot P, Carella F, Toscano A, Soliveri P, Girlanda P, Carrara F, Fratta GM, Reid FM, Mariott C, Zeviani M. Maternally inherited hearing loss, ataxia and myoclonus associated with a novel point mutation in mitochondrial tRNAser(ucn) gene. Hum. Mol. Genet., 1995;4:1421-7.
21. Pavlakis SG, Phillips PC, Dimauro S, De Vito DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes: a distinctive clinical syndrome. Ann. Neurol., 1984;16:481-8.
22. Zeviani M, Tiranti V, Piantadosi C. Mitochondrial disorders. Medicine, 1998;77:59-72.
23. Maassen JA & Kadowaki T. Maternally inherited diabetes and deafness: a new diabetes subtype. Diabetologia, 1996;39:375-82.
24. Lindsay JR, Hinojosa R. Histopathologic features of the inner ear associated with Kearns-Sayre syndrome. Arch. Otolaryngol., 1976;102:747-52.
25. Hammans SR, Sweeney MG, Hanna MG, Brockington M, Morgan-Hughes JA, Harding AE. The mitochondrial DNA transfer RNALEU(UUR) A®G(3243) mutation: a clinical and genetic study. Brain, 1995;118:721-34.
26. Vernham GA, Reid FM, Rundle PA, Jacobs HT. Bilateral sensorineural hearing loss in members of a maternal lineage with a mitochondrial point mutation. Clin. Otolaryngol., 1994;19:314-9.
27. Rosenthal EL, Kileny PR, Boerst A, Telian SA. Successful cochlear implantation in a patient with MELAS syndrome. Am. J. Otol., 1999;20:187-91.
28. Tono T, Ushisako Y, Kiyomizu K, Usami S, Abe S, Shinkawa H, Komune S. Cochlear implantation in a patient with profound hearing loss with the A1555G mitochondrial mutation. Am. J. Otol., 1998;19:754-7.
29. Yamaguchi T, Himi T, Harabuchi Y, Hamamoto M, Kataura A. Cochlear implantation in a patient with mitochondrial disease - Kearns-Sayre syndrome: a case report. Adv. Otorhinolaryngol., 1997;52:321-3.
1 Ph.D., Department of Otorhinolaryngology, Medical Sciences School, Santa Casa de São Paulo.
2 Joint Professor, Department of Otorhinolaryngology, Medical Sciences School, Santa Casa de São Paulo.
Study conducted at the Department of Otorhinolaryngology, Santa Casa de São Paulo. Master Dissertation thesis presented to the Post-Graduate Course of Medical Sciences School, Santa Casa de São Paulo.
Address correspondence to: Dra. Maria de Fátima Carvalho - Rua Hilário Furlan 107 - Brooklin Novo - CEP 04571-180 São Paulo / SP. - Tel/Fax: (55 11) 5505-1915 - E-mail fatimaorl@uol.com.br
Article submitted on March 7, 2001. Article accepted on April 3, 2001.

Print: ![]()