

Year: 2003 Vol. 69 Ed. 2 - (19º)
Relato de Caso
Pages: 267 to 271
 PDF PT
PDF PT Morquio's syndrome: case report and review
Author(s):
Adriana G. Chaves[1],
Karina B. Tavares1,
José R. Val[2],
Cícero Matsuyama[3],
Paulo E. Riskalla[4]
Keywords: Morquio's syndrome, mucopolysaccharidosis, auditive deficiency.
Abstract:
Introduction: Morquio's Syndrome is a mucopolysaccharidosis type IV commomnly associated with auditive deficiency, although this association is not well described yet. Objective: To evaluate the finding of auditive deficiency in a patient with Morquio's Syndrome. Methods: We describe the case of a 16-year-old male patient, with the diagnosis of Morquio's Syndrome, that underwent physical examination and audiometric evaluation to determine the auditive loss. Results: The patient had a history of skeletal complaints and since he was 2 years old. His parents were related and there was a positive family history for the same findings. For the last five years he has been complaining of progressive auditive loss. He had a typical appearence and weighted 27 Kg, with a stature of 99 cm, short neck, globous thorax and abdomen, genu valgum and wrists hyperextension. Otoscopy showed bilateral opacification of the ear drums and audiometry revealed mild conductive disacusis. Discussion: Mucopolysaccharidosis (MPS) are caused by the deficiency of liposomal enzymes, leading to progressive accumulation of mucopolysaccharhides into the tissues. Morquio's Syndrome is a MPS type IV inherited in a autosomic recessive pattern. Tissue deposits may occur into the visceral organs, bones, cornea and ears leading to structural and functional abnormalities. Conclusions: Morquio's Syndrome is a metabolic disease of carbohydrates usually beginning between 18 and 24 months with a variable range of clinical findings. It is important to make the diagnosis early in order to avoid the limitations of the sensory losses.
![]()
INTRODUCTION
Morquio's syndrome was initially and simultaneously described as dwarfism with bone malformations by Morquio (Uruguayan pediatrician) and Ulfrich (English radiologist) in 19291,2,3.
It is a hereditary osteochondrodystrophy characterized by connective tissue affection, implying congenital metabolism error of polysaccharides, especially of keratosulfate (type IV mucopolysaccharide). Its transmission is autosomal recessive, affecting approximately 1 in each 40,000 births1, 2, 4-9. If affects both genders similarly3 and occurs more frequently in consanguineous marriages of healthy heterozygote parents2, 3.
The described etiology is N-acetyl-galactosamine 6 sulfatase deficiency, leading to accumulation of polysaccharides. The patients are apparently normal at birth and present normal neural psychomotor development, with preserved intelligence throughout the whole life3.
Clinical abnormalities are detected at 18-24 months of age, with significant height and weight delay. Among the typical skeletal abnormalities we can include dwarfism with short trunk, pectus carinatum, kyphosis, hyperlordosis, scoliosis, vertebral ovoid deformity, genus valgum, bilateral valgum flat foot, joint hyperextension (especially of the wrists), and odontogenic hypoplasia. Among the extra-skeletal manifestations we can include: opacity of cornea, hepatomegalia (prominent abdomen), cardiac valve damage, prognathism, short and flat nose, spaced teeth and reduction of dental enamel, and frequent hearing loss, which can range from conductive to sensorineural losses1-4, 6-10.
For diagnosis, the following tools are important: ophthalmic test with light, urine chromatography to detect the dosage of keratosulfate (which presents high levels in the syndrome), radiological study of the neck, dorsal, lumbo-sacral spine and limbs, specific enzyme study in tissues, fibroblasts and leukocytes, and audiometry associated with immittance measures1-4, 6, 8, 10, 11.
Since these patients have normal psychomotor development, it is essential to make an early diagnosis, so that the appropriate orthopedic treatment and effective rehabilitation can be provided, together with genetic counseling.
CASE REPORT
WCO, male 16-year old patient, come to the ambulatory complaining of progressive bilateral hearing loss for approximately 5 years. He had had an uneventful normal delivery at home. He did not report preexisting diseases (patient had never seen a doctor before). Parents were consanguineous relatives (first-degree cousins). He had 9 siblings (six sisters and three brothers), and one brother had died at the age of 21 years, with a similar condition. He reported three cousins who shared the same physical characteristics (two women, one of which had died at the age of 25 owing to respiratory complications and one man).
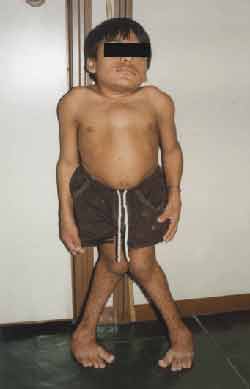
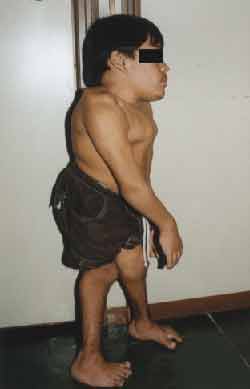
Physical examination: Height: 99 centimeters, Weight, 27 kilos. (Figures 1 and 2)
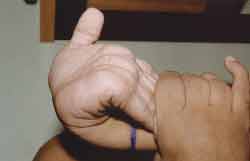
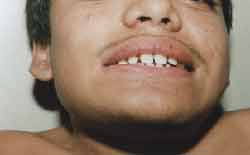
Trunk and neck were short, increase in anterior-posterior thorax diameters, with pectus carinatum shape, genus valgum, adduction feet, could not stay long in orthostatic position, hyperextension of wrists (Figure 3) and ankles, globous abdomen and hepatospleenomegalia. Pulmonary and heart auscultation within normal range. ENT examination: oroscopy - spaced and yellowish teeth (Figure 4). Otoscopy: opacification of tympanic membrane bilaterally.
The patient also saw the ophthalmology that detected mild corneal opacification.
We conducted cardiac tests, such as electrocardiogram and echocardiogram, which were within the normal range. We ordered pure tone and speech audiometry. Pure tone audiometry demonstrated mild conductive hearing loss on the right with air-bone gap of 10dB. Speech audiometry presented 92% discrimination bilaterally and SRT (right ear: 35dB and left ear: 25dB). Left ear immitance measures did not show abnormalities, whereas the left ear presented negative pressure at the level of -300daPa. The stapedial acoustic reflex evidenced absence on the right ear and reduction on the left ear (Figure 5).
Figure 1. Height abnormalities (anterior-posterior).
Figure 2. Height abnormalities (profile).
Figure 3. Wrist hyperextension.
Figure 4. Spaced teeth.
Figure 5. Audiometry and Immittance measures.
DISCUSSION
Mucopolysaccharidosis (MPS) encompasses a group of diseases caused by deficiency in liposomal enzymes involved in the metabolism of mucopolysaccharides 5, 6, 8. As a result, the non-degraded mucopolysaccharides are accumulated intracellularly, originating large cells with cytoplasmatic nuclei and determining cell function abnormalities8. MPS are progressive disorders characterized by involvement of multiple organs: liver, spleen, heart, vessels, bone marrow, lymph nodes, osteoarticular system, eyes and ears8. Currently, ten enzyme deficiencies were identified and classified into 7 types or syndromes5, 8.
Morquio's syndrome is a type IV MPS genetically transmitted (autosomal recessive). The basic defect consists of an enzymatic deficiency: reduction of N-acetyl galactosamine 6 sulfatase (Type IV A), leading to incomplete degradation of mucopolysaccharides and consequent tissue deposition with progressive lesion5, 6, 8, 9. It is characterized by achondroplastia, osteochondrodysplasia and keratosulfaturia. It is associated with chronic otitis media and mixed or sensorineural hearing loss9, 12. The main differential diagnosis should be made with rickets and other MPS forms, especially Hurler's syndrome (MPS type I), which is differentiated by the presence of mental retardation1, 2, 4, 5, 9, 10. Auditory abnormalities are not constant and normally its onset is after 10 years of age3, ranging from normal hearing to anacusis. In a study conducted with 18 patients at Johns Hopkins Hospital, 17 patients presented hearing loss, in which 14 had history of otitis media. Patients aged below 8 years presented conductive loss, possibly owing to a biochemical abnormality of the fundamental substance of the ossicle chain, leading to bone deformity. Older patients presented sensorineural or mixed loss, secondary to excessive deposition of mucopolysaccharide in the cochlear region. In all of them, except for three patients aged over 8 years, the hearing loss was mild to moderate7, 13. According to Cummings, the conductive component can be attributed to secondary serous otitis media, Eustachian tube dysfunction, and chronic thickness of middle ear mucosa14. Clinical pathology description of the temporal bones confirmed the existence of mucopolysaccharide deposits in the middle ear and vascular stria 7, in addition to the presence of hemorrhage among the collagen fibers of the external acoustic meatus, facial canal and around the ossicle chain13. The patients normally develop aorta failure, tendency to bleeding and ventilation disorders, secondary to bone marrow compression6, 9, 10.
We conducted urine chromatography that showed high levels of keratosulfate. The radiological study of the cervical, dorsal, lumbo-sacral spine and limbs demonstrated skeletal abnormalities compatible with the syndrome. Ophthalmologic and audiological exams observed in the patient in question conducted to the characterization of Morquio's syndrome. The disease can occur up to adult life, but it is normally stabilized at adolescence. If the patient has stable disease, especially the cardiac function, survival can exceed 40 years.
CLOSING REMARKS
Morquio's syndrome is a disease that causes severe clinical affections. The later the diagnosis, the greater the impairment of the locomotion function. Since it may be associated with hearing loss, we should investigate hearing status as early as possible, so that there are no communication development impairments.
REFERENCES
1. Canto RS et al. Osteocondrodistrofia deformante (Doença de Morquio): estudo de uma família. Revista Brasileira de Ortopedia 1986;21(1):16-22.
2. Mena M, Obando R. Síndrome de Morquio. Revista Chilena de Pediatria 1976;47(3):247-253.
3. Redondo MR, Barri TS, Belmonte DS. Datos diagnósticos en la enfermedad de Morquio. An Esp de Pediatr 1987;26(6):471-472.
4. Beck M et al. Heterogeneity of Morquio disease. Clinical Genetics 1986;29: 325-331.
5. Fitzgerald J, Verveniotis SJ. Morquio's Syndrome. A case report and review of clinical findings. N Y State Dent J 1998;64(8):48-580.
6. Goldman L, Bennet JC. Cecil Textbook of Medicine. 21st Edition; 1999. p. 1116-1118.
7. Riedner ED, Levin LS. Hearing Patterns in Morquio's Syndrome (Mucopolysaccharidosis IV). Arch Otolaryngol 1977;103, 518-520.
8. Robbins S, Kumar V. Pathologic basis of disease. W. B. Saunders Company; 1994. p. 114-115.
9. Sataloff RT, Schiebel BR, Spiegel JR. Morquio's Syndrome. The American Journal of Otology 1987;8(5):443-449.
10. Moreno Y et al. Manejo anestésico en el Síndrome de Morquio. A propósito de un caso? Revista Venezoelana de Anestesiología 1999;4(1):26-29.
11. Ipinza IH, Claver F. Sindrome de Morquio. Reportaje de un caso clinico-radiologico. Revista Médica Del Maule 1986;5(1):131-5.
12. Cruz OLM, Costa SS. Otologia Clínica e Cirúrgica. 1ª edição. Rio de Janeiro; 2000.
13. Kelemen G. Morquio's Disease and the Hearing Organ. ORL 1977;39: 233-240.
14. Cummings CW et al. Otolaryngology Head & Neck Surgery. 3rd edition; 1998. p. 1374.
Print: ![]()