

Ano: 2003 Vol. 69 Ed. 2 - Março - Abril - (19º)
Seção: Relato de Caso
Páginas: 267 a 271
 PDF PT
PDF PT Síndrome de Morquio: relato de caso e revisão da literatura
Morquio's syndrome: case report and review
Autor(es):
Adriana G. Chaves[1],
Karina B. Tavares1,
José R. Val[2],
Cícero Matsuyama[3],
Paulo E. Riskalla[4]
Palavras-chave: síndrome de Morquio, mucopolissacaridose, perda auditiva.
Keywords: Morquio's syndrome, mucopolysaccharidosis, auditive deficiency.
Resumo:
Introdução: A Síndrome de Morquio é uma mucopolissacaridose tipo IV, geralmente associada a perda auditiva. Objetivo: Investigar a presença de deficiência auditiva em paciente com a Síndrome de Morquio. Material e Método: Analisamos um paciente do sexo masculino, 16 anos, sindrômico, com alterações esqueléticas e extra-esqueléticas desde os dois anos de idade. Pais consangüíneos e quadro semelhante na família. Paciente apresentava queixa de hipoacusia progressiva há cerca de cinco anos. Resultados: Paciente com estatura = 99 cm, peso = 27 kg, pescoço curto, aumento do diâmetro ântero-posterior, abdome globoso, geno valgo e hiperextensão de punhos. A otoscopia evidenciou opacificação bilateral das membranas timpânicas e o exame audiométrico demonstrou disacusia condutiva, de leve intensidade. Discussão: As mucopolissacaridoses (MPS) são doenças causadas pela deficiência de enzimas lisossomais, resultando em acúmulo de mucopolissacarídeos em diversos tecidos corporais. A Síndrome de Morquio é uma MPS do tipo IV, transmitida por herança autossômica recessiva. Os depósitos tissulares podem se localizar nas vísceras, ossos, córnea e ouvidos, desencadeando alterações estruturais e funcionais. Conclusão: A Síndrome de Morquio é uma doença do metabolismo dos carboidratos, manifestando-se a partir dos 18 a 24 meses de idade e apresentando um espectro de manifestações clínicas. Ao otorrinolaringologista cabe detectar precocemente perdas auditivas, a fim de minimizar as limitações da própria evolução da doença.
Abstract:
Introduction: Morquio's Syndrome is a mucopolysaccharidosis type IV commomnly associated with auditive deficiency, although this association is not well described yet. Objective: To evaluate the finding of auditive deficiency in a patient with Morquio's Syndrome. Methods: We describe the case of a 16-year-old male patient, with the diagnosis of Morquio's Syndrome, that underwent physical examination and audiometric evaluation to determine the auditive loss. Results: The patient had a history of skeletal complaints and since he was 2 years old. His parents were related and there was a positive family history for the same findings. For the last five years he has been complaining of progressive auditive loss. He had a typical appearence and weighted 27 Kg, with a stature of 99 cm, short neck, globous thorax and abdomen, genu valgum and wrists hyperextension. Otoscopy showed bilateral opacification of the ear drums and audiometry revealed mild conductive disacusis. Discussion: Mucopolysaccharidosis (MPS) are caused by the deficiency of liposomal enzymes, leading to progressive accumulation of mucopolysaccharhides into the tissues. Morquio's Syndrome is a MPS type IV inherited in a autosomic recessive pattern. Tissue deposits may occur into the visceral organs, bones, cornea and ears leading to structural and functional abnormalities. Conclusions: Morquio's Syndrome is a metabolic disease of carbohydrates usually beginning between 18 and 24 months with a variable range of clinical findings. It is important to make the diagnosis early in order to avoid the limitations of the sensory losses.
![]()
INTRODUÇÃO
A Síndrome de Morquio foi descrita inicialmente como um nanismo com malformações ósseas, simultaneamente por Morquio (pediatra uruguaio) e Ulfrich (radiologista inglês), em 19291,2,3.
É uma osteocondrodistrofia hereditária que se caracteriza por um transtorno do tecido conjuntivo, implicando em um erro congênito do metabolismo dos polissacarídeos, em particular dos queratossulfatos (mucopolissacaridose tipo IV). Sua transmissão é autossômica recessiva, ocorrendo aproximadamente em 1 em cada 40. 000 nascimentos1,2,4-9. Acomete ambos os sexos da mesma maneira3 e ocorre, com maior freqüência, em casamentos consangüíneos de pais sadios e heterozigotos2,3.
A etiologia descrita seria a deficiência da enzima N-acetil galactosamina 6 sulfatase, levando a um acúmulo de mucopolissacarídeos. Os pacientes são aparentemente normais ao nascimento e apresentam desenvolvimento neuro-psicomotor normal com a inteligência preservada durante toda a vida3.
As alterações clínicas começam a ocorrer a partir dos 18 a 24 meses de idade, com a instalação de grande retardo pondo-estatural. Dentre as alterações esqueléticas típicas pode ocorrer: nanismo com tronco curto, tórax em quilha (pectus carinatum), cifose, hiperlordose, escoliose, deformidade ovóide das vértebras, geno valgo, pé plano valgo bilateral, hiperextensão articular (principalmente dos punhos) e hipoplasia odontogênica. Dentre as manifestações extra-esqueléticas pode haver: opacidade da córnea, hepatomegalia (abdome proeminente), lesões valvulares cardíacas, prognatismo, nariz curto e achatado, dentes espaçados e com redução no esmalte, e frequentemente alterações auditivas, que podem variar desde disacusias condutivas até neurossensoriais1-4,6,-10.
São de utilidade para o diagnóstico o exame oftalmológico com lâmpada de fenda, cromatografia da urina a fim de detectar a dosagem de queratossulfato (que se acha em níveis elevados na Síndrome), estudo radiológico de coluna cervical, dorsal, lombossacral e de extremidades, a pesquisa enzimática específica nos tecidos, fibroblastos e leucócitos e a audiometria associada a imitanciometria1-4,6,8,10,11.
Como estes pacientes apresentam um desenvolvimento psicomotor normal é imprescindível um diagnóstico precoce, para que sejam estabelecidos tratamento ortopédico e reabilitação eficaz, assim como um aconselhamento genético.
CASO CLÍNICO
A. W. C. O., sexo masculino, pardo, 16 anos de idade, procurou atendimento em nosso ambulatório queixando-se de hipoacusia progressiva bilateral há aproximadamente cinco anos. Nasceu de parto normal domiciliar sem intercorrências. Nega doenças pré-existentes (paciente nunca havia se consultado com qualquer outra especialidade médica). Pais consangüíneos (primos de primeiro grau). Possui nove irmãos (seis mulheres e três homens), sendo que um irmão faleceu aos 21 anos de idade, com quadro clínico semelhante. Também relata três primos com as mesmas características físicas (duas mulheres, uma das quais evolui para óbito aos 25 anos, devido a complicações respiratórias, e um homem).
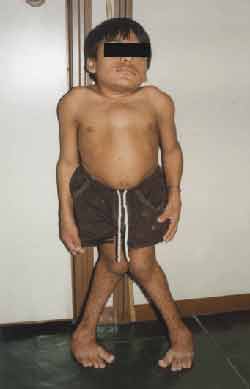
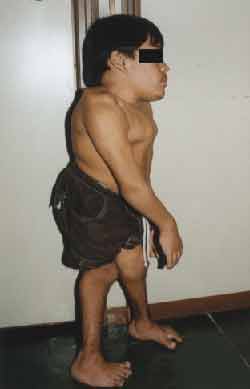
Ao exame físico: Estatura, 99 centímetros, Peso, 27 quilos. (Figuras 1 e 2)
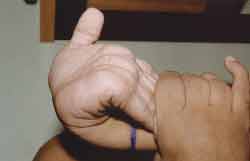
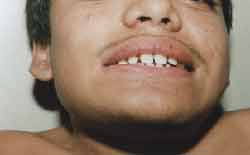
Apresenta tronco e pescoço curtos, aumento do diâmetro ântero-posterior do tórax, com formato de peito de pombo, geno valgo, pés em adução, não se mantendo por muito tempo em posição ortostática, hiperextensão de punhos (Figura 3) e tornozelos, abdome globoso e hepatoesplenomegalia. Auscultas pulmonar e cardíaca, sem alterações. Exame otorrinolaringológico: oroscopia: dentes espaçados e amarelados (Figura 4). Otoscopia: opacificação de membranas timpânicas bilateralmente.
O paciente passou em consulta com oftalmologista o qual evidenciou uma opacificação leve de córneas.
Foram realizados exames cardiológicos como eletrocardiograma e ecocardiograma, os quais resultaram normais. Solicitamos a audiometria tonal e vocal. A audiometria tonal demonstrou hipoacusia condutiva de grau leve em orelha direita com diferença aéreo-óssea de 10 dB. A logoaudiometria apresentou discriminação de 92% bilateralmente e SRT (orelha direita: 35 dB e orelha esquerda: 25 dB). A imitanciometria da orelha esquerda não mostrou alteração, enquanto a da orelha esquerda apresentou pressão negativa ao nível de -300 daPa. A pesquisa do reflexo do músculo estapédio evidenciou ausência em orelha direita e diminuição em orelha esquerda (Figura 5).
Figura 1. Alterações estaturais (ântero-posterior)
Figura 2. Alterações estaturais (perfil)
Figura 3. Hiperextensão de punhos
Figura 4. Dentes espaçados
Ver figura
Figura 5. Audiometria e Imitanciometria
DISCUSSÃO
As mucopolissacaridoses (MPS) são um grupo de doenças causadas por uma deficiência nas enzimas lisossomais envolvidas no metabolismo dos mucopolissacarídeos5,6,8. Como resultado, esses mucopolissacarídeos não degradados se acumulam intracelularmente, originando células grandes com vacúolos citoplasmáticos e determinando alterações da função celular8. As MPS são desordens progressivas, caracterizadas pelo envolvimento de múltiplos órgãos: fígado, baço, coração, vasos sanguíneos, medula óssea, linfonodos, sistema osteoarticular, olhos e ouvidos8. Atualmente, dez deficiências enzimáticas foram identificadas e classificadas em 7 tipos ou síndromes5,8.
A Síndrome de Morquio é uma mucopolissacaridose do tipo IV, geneticamente transmitida (herança autossômica recessiva). O defeito básico consiste em uma deficiência enzimática: diminuição da N-acetilgalactosamina 6-sulfatase (tipo IV A), levando à incompleta degradação dos mucopolissacarídeos e conseqüente deposição tissular com lesão progressiva5,6,8,9. É caracterizada por acondroplasia, osteocondrodisplasia e queratosulfatúria. Associa-se à otite média crônica e disacusia mista ou neurossensorial9,12. Os principais diagnósticos diferenciais devem ser o raquitismo e as outras mucopolissacaridoses (MPS), principalmente a Síndrome de Hurler (MPS tipo I), que se diferencia pela presença de retardo mental1,2,4,5,9,10. As alterações auditivas não são constantes e geralmente aparecem após os dez anos de idade3, variando desde audição normal até anacusia. Em estudo realizado com 18 pacientes no Johns Hopkins Hospital, 17 demonstraram perda auditiva, sendo que 14 tinham história de otite média. Pacientes com idade inferior a oito anos apresentaram perda do tipo condutiva, possivelmente devido a uma alteração bioquímica na substância fundamental da cadeia ossicular, conduzindo a deformidade óssea. Pacientes maiores possuíam perda neurossensorial ou mista, secundária a deposição dos mucopolissacarídeos em excesso na região coclear. Em todos, exceto em três pacientes acima de oito anos, a perda auditiva era discreta ou moderada7,13. Segundo Cummings, o componente condutivo pode ser atribuído à otite média serosa secundária, à disfunção da tuba de Eustáquio e ao espessamento crônico da mucosa da orelha média14. Descrições anatomopatológicas de ossos temporais comprovam a existência de depósitos de mucopolissacarídeos na orelha média e estria vascular7, além da presença de hemorragias entre as fibras colágenas no meato acústico externo, canal do facial e ao redor da cadeia ossicular13. Os pacientes geralmente desenvolvem insuficiência aórtica, tendência a sangramentos e distúrbios ventilatórios, secundários a compressão medular6,9,10.
Foi realizada cromatografia da urina que evidenciou níveis elevados de queratossulfato. O estudo radiológico de coluna cervical, dorsal, lombo-sacral e de extremidades demonstrou alterações esqueléticas compatíveis com a síndrome. Os exames oftalmológicos e audiológicos observados no paciente em questão conduzem a caracterização da Síndrome de Morquio. A doença pode ocorrer até a vida adulta, mas, geralmente estabiliza-se na adolescência. Estando o paciente com a doença estabilizada e, principalmente a função cardíaca controlada, a sobrevida poderá ser superior a 40 anos.
COMENTÁRIOS FINAIS
A Síndrome de Morquio é uma doença que causa graves alterações clínicas. Quanto mais tardio o diagnóstico, maior será o comprometimento da função locomotora. Como podem ocorrer alterações auditivas nestes pacientes, deve-se pesquisar sua audição o mais precocemente possível, para que não ocorram prejuízos ao desenvolvimento de comunicação.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Canto RS et al. Osteocondrodistrofia deformante (Doença de Morquio): estudo de uma família. Revista Brasileira de Ortopedia 1986;21(1):16-22.
2. Mena M, Obando R. Síndrome de Morquio. Revista Chilena de Pediatria 1976;47(3):247-253.
3. Redondo MR, Barri TS, Belmonte DS. Datos diagnósticos en la enfermedad de Morquio. An Esp de Pediatr 1987;26(6):471-472.
4. Beck M et al. Heterogeneity of Morquio disease. Clinical Genetics 1986;29: 325-331.
5. Fitzgerald J, Verveniotis SJ. Morquio's Syndrome. A case report and review of clinical findings. N Y State Dent J 1998;64(8):48-580.
6. Goldman L, Bennet JC. Cecil Textbook of Medicine. 21st Edition; 1999. p. 1116-1118.
7. Riedner ED, Levin LS. Hearing Patterns in Morquio's Syndrome (Mucopolysaccharidosis IV). Arch Otolaryngol 1977;103, 518-520.
8. Robbins S, Kumar V. Pathologic basis of disease. W. B. Saunders Company; 1994. p. 114-115.
9. Sataloff RT, Schiebel BR, Spiegel JR. Morquio's Syndrome. The American Journal of Otology 1987;8(5):443-449.
10. Moreno Y et al. Manejo anestésico en el Síndrome de Morquio. A propósito de un caso? Revista Venezoelana de Anestesiología 1999;4(1):26-29.
11. Ipinza IH, Claver F. Sindrome de Morquio. Reportaje de un caso clinico-radiologico. Revista Médica Del Maule 1986;5(1):131-5.
12. Cruz OLM, Costa SS. Otologia Clínica e Cirúrgica. 1ª edição. Rio de Janeiro; 2000.
13. Kelemen G. Morquio's Disease and the Hearing Organ. ORL 1977;39: 233-240.
14. Cummings CW et al. Otolaryngology Head & Neck Surgery. 3rd edition; 1998. p. 1374.
[1] Médicas Residentes de Otorrinolaringologia do Hospital Cema, São Paulo.
[2] Médico Otorrinolaringologista do Hospital Cema, São Paulo.
[3] Coordenador Geral da Residência Médica do Hospital Cema. Mestre e Doutorando em Otorrinolaringologia pela UNIFESP-EPM.
[4] Professor Adjunto UNIFESP-EPM.
Hospital Cema - São Paulo, SP.
Endereço para correspondência: Adriana G. Chaves - R. Madre de Deus, 1361 apto. 52 Mooca São Paulo SP - Tel (0xx11)94523250 - E-mail: Adrichaves@bol.com.br
Imprimir: ![]()