

Year: 2008 Vol. 74 Ed. 2 - (25º)
Relato de Caso
Pages: 307 to 314
 PDF PT
PDF PT  PDF EN
PDF ENRare nasosinusal tumors: Case series and literature review
Author(s): Roberta de Paula Araújo 1, Érika Ferreira Gomes 2, Dalgimar Beserra de Menezes 3, Lidiane Maria de Brito Macedo Ferreira 4, Adson Sales do Nascimento Rios 5
Keywords: nasal cavity, neoplasm
Abstract:
Tumors of the nasal cavity and paranasal sinuses are unusual pathologies found in clinical practice. Approximately 0.8% of all human cancers are located in this area. Despite being rare, nasosinusal neoplasms usually manifest through nonspecific symptoms that are common to numerous inflammatory pathologies. The aim of this study is to describe a series of rare nasosinusal tumors, including esthesioneuroblastomas, central giant cell granulomas, extramedullary plasmocytomas, nasosinusal hemangiopericytomas, neurofibromas and cemento-ossifying fibromas, diagnosed at the Fortaleza General Hospital. We, hereby, briefly review each of the aforementioned pathologies, stressing the need for a precise histological diagnosis for proper treatment in each case.
![]()
INTRODUCTION
Nasosinusal malignant tumors are rare, representing less than 3% of head and neck cancers and 0.8% of all human cancers1. Approximately 55% originate in the maxillary sinuses, 35% in the nasal cavity, 9% in the ethmoid and 1% in the frontal and sphenoid sinuses2. In the USA, the incidence of nasal cavity tumors is less than 1 case in 100 thousand persons per year2. With the exception of non-epithelial tumors, nasosinusal cancer is a disease that affects adults, being more frequent in men above 50 years of age2.
Tumors in this region usually cause unspecific and common symptoms, including inflammatory diseases. Nasal obstruction (61.2%), epistaxis (40.8%), facial pain (39.2%) and local infection (23.9%) are the most commonly reported initial symptoms1. Such fact, associated with the low incidence of these tumors and, often times, the difficulties associated with histopathology, contribute to delays in diagnosis and treatment.
CLINICAL CASES
Case 1
ATS, female, 16 years, coming from Acarape/CE, came to us complaining of epistaxis followed by right side nasal obstruction for 3 years. Anterior rhinoscopy showed a hyperemic tumoral lesion, brittle, involving the upper portion of the right nasal cavity (RNC), extending all the way to the middle meatus.
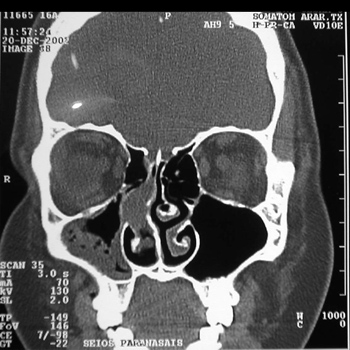
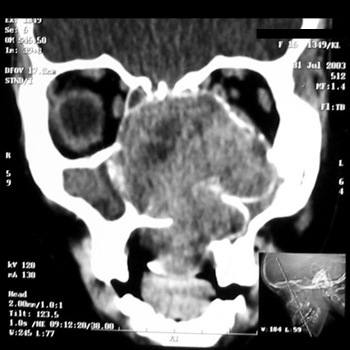
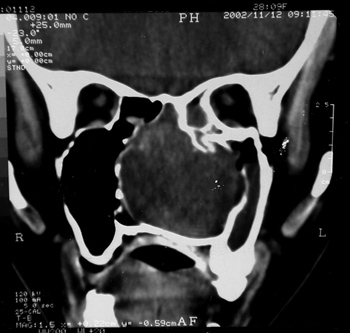
Nasal cavity and paranasal sinuses CT scan showed a dense polypoid mass, well defined, in the right nasal cavity, fully obliterating the middle meatus and the spheno-ethmoidal recess, with middle turbinate and poorly defined ipsilateral posterior ethmoidal air cells. We could also see a thickening of the right maxillary and sphenoidal sinuses mucosa (Figure 1).
Figure 1 - CT scan. Coronal view of a Stesioneuroblastoma (case 1).
Incision biopsy followed by nasal packing was carried out in the outpatient ward. Pathology exams showed clusters of small cells of round hyperchromatic nuclei amidst fibril stroma, matching signs of stesioneuroblastoma. Having a diagnosis of Kadish B (nasal cavity and paranasal sinuses involvement), we proposed a mid-facial degloving resection, followed by radiotherapy (RxT). Still during the preoperative, the patient had intensive epistaxis, requiring arteriography and selective right maxillary artery catheterization for tumor embolization.
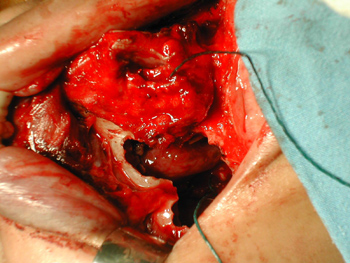
In the eighth post-embolization day, we carried out a cranio-facial tumor resection. The lesion occupied the upper portion of the right nasal cavity, measuring approximately 4cmx3cmx1.5cm. Anterior maxillary sinusectomy showed no tumoral mass in the right maxillary sinus (Figure 2). The medial wall of the maxillary sinus was resected and the ethmoid lamina cribosa was drilled all the way to the dura-mater, to guarantee surgical mass removal. The patient was then referred to RxT, and today is in the 8th month of follow up, without signs of recurrence.
Figure 2 - Middle-face degloving with anterior maxillary sinusotomy, showing stesioneuroblastoma (case 1).
Case 2
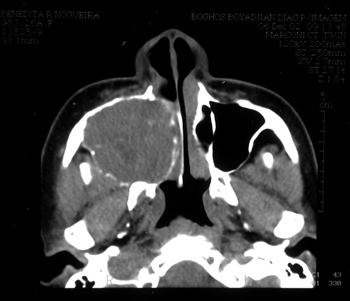
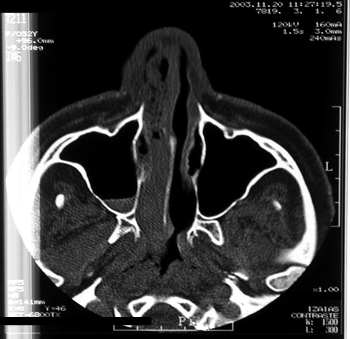
BRN, Female, 26 years, came from Fortaleza/CE, came to us complaining of a bulging on the hemiface and right nasal obstruction, with 8 months of development. She reported she had a tooth removed (1st right pre-molar) one year before. Her CT scan showed an expansive solid large heterogeneous mass occupying her right maxillary sinus, with expansion and small areas of cortical invasion. The lesion extended to the hard palate and ipsilateral tooth alveolar process, to the RNC and partially obstructed the inferior orbital fissure (Figure 3).
Figure 3 - CT scan - axial cross section of a GCCG (case 2).
When we biopsied her through a sublabial antrostomy, we could see no anterior bone wall on the right maxillary sinus and a brittle encapsulated bulky and vascularized tumor. An AP study showed tumor sections reproducing giant multinucleated cells inserted in the fibrous stroma of variable density, with ovoid and fusiform cells proliferation, in a clinical picture matching that of giant cells central granuloma (GCCG). Treatment proposed was direct maxillectomy through mid-facial degloving. The patient is in her 24th month of follow up, with no evidences of tumor recurrence (Figure 4).
Figure 4 - CT scan - coronal view of a GCCG post operative (case 2).
Case 3
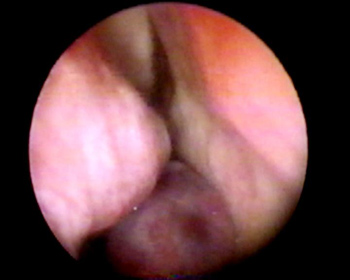
CAS, female, 52 years, coming from Fortaleza/CE, complained of epistaxis for 3 years, followed by right side nasal obstruction. Her otorhinolaryngological physical exam was normal. Through nasal-endoscopy we could see a tumor on the RNC floor, extending to the nasopharynx (Figure 5). Paranasal sinuses CT scan showed an expansive homogeneous mass, of soft tissue density in her right nasal cavity and nasopharynx, without evidence of bone erosion (Figure 6). We carried out an incisional biopsy, showing connective and vascular neoformation, with intense infiltration of plasmocytic cells and respiratory-type cell, matching signs of plasmocitic granuloma. We ordered lab exams in order to rule out systemic disease (multiple myeloma). Blood tests, bone marrow aspirate and biopsy, together with protein electrophoresis were all within normal ranges. The tumor was excised by endoscopy, with no evidences of bone invasion or that of adjacent structures. The patient was referred to complementary RxT, and is in her 8th month of follow up, without evidences of tumor recurrence or systemic disease.
Figure 5 - RNC Extramedullary Plasmocytoma - nasal fibroscopic view. (case 3).
Figure 6 - CT scan - axial view of Extramedullary Plasmocytoma (case 3).
Case 4
LDS, 15 years, female, coming from Fortaleza/CE, came to us complaining of tooth ache and bolus lesion on her palate to the left, followed by epistaxis, hemifacial bulging and ipsilateral exophthalmus, of about one year of evolution. Paranasal sinuses CT scan showed an expansive mass in the left nasal cavity, extending anteriorly to the orbit apexes and supra-sellar region, destroying the pterygoid lamina. The lesion expanded to the ipsilateral sphenoidal and maxillary sinuses and to the hard palate, uptaking little contrast. (Figure 7).
Figure 7 - CT scan - coronal view of a nasosinusal hemangiopericytoma (case 4).
At our first biopsy attempt, there was an important bleeding, then we chose to do an arteriography and tumor embolization, which presented expressive regression. We did an excision biopsy through anterior maxillary antrostomy, and we only saw the tumoral capsule and the necrotic tissue in a large cavity within the maxillary sinus.
AP study showed connective tissue with mixed infiltrate of inflammatory cells and vascular clefts with collarette of cells with round nuclei; areas of mucinous material build up and deposits of hemosiderin pigment. We concluded it was an angiomatoid tumor, with no signs of malignancy, with a differential diagnosis between glomangioma and nasosinusal hemangiopericytoma. Tumoral cells laying around vascular spaces that showed only vimentin in the immunohistochemistry exam, negative for actin, smooth muscle actin, CD31 and CD34, is more compatible with nasosinusal hemangiopericytoma.
Three months after the surgical procedure, the patient returned complaining of epistaxis. During nasal-endoscopy we noticed a residual tumor in her RNC floor, extending to the soft palate. CT scan showed a lesion measuring about 5cm X 3cm, with bone destruction and rarefaction, involving the orbital floor, lateral wall and maxillary sinus floor. The lesion was destroying the clivus, ethmoid and sphenoid sinuses on the left side, invading the parasellar region and involving the cavernous sinus (Figure 8).
Figure 8 - CT scan - axial view of a relapsed nasosinusal hemangiopericytoma (case 4).
The patient underwent RxT and chemotherapy (QT), and died about one year after the diagnosis, with clinical signs of intracranial hemorrhage.
Case 5
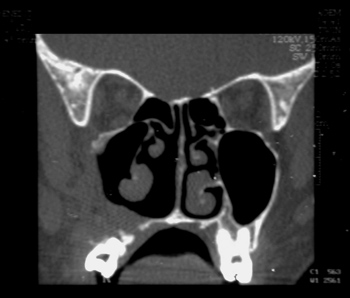
AAGL, female, 23 years old, coming from Maranguape/Ce, with previous history of epistaxis and left side nasal obstruction for 5 years. She had a hyperemic and non-bleeding tumor in her LNC, and ipsilateral bulging on the nose, maxillary and palate regions. Her CT scan showed an expansive lesion in her left nasal cavity (LNC), occupying the rhinopharynx and ipsilateral maxillary and ethmoidal sinuses, with right side nasal septum deviation and no signs of bone lysis (Figure 9).
Figure 9 - CT scan - coronal view of a neurofibroma (case 5).
She underwent an incisional biopsy, which showed proliferation of elongated and fusiform cells, some with nuclei arranged in bundles, matching signs of neurofibroma. We carried out a sublabial maxillary antrostomy to resect the lesion, which measured approximately 9cmx9cmx2cm. She is being followed up for 17 months now, and there is no evidence of tumor relapse.
Case 6
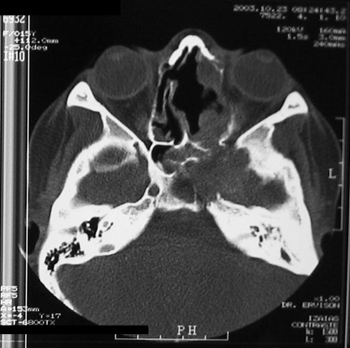
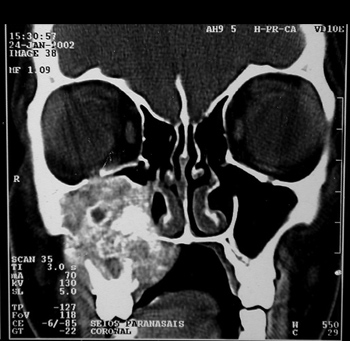
MVPM, female, 15 years old, had a bulging in her right infraorbital region, periodontal ulceration of the upper molars and submucosal bulging of her ipsilateral hard palate that had been evolving from one year. She did not have epistaxis or nasal obstruction. CT scan showed an expansive lesion with concentric calcifications in her right-side maxillary sinus, destroying its walls and invading the hard palate (Figure 10).
Figure 10 - CT scan - coronal view of a cement-ossifying fibroma (case 6).
We carried out an incisional biopsy through anterior maxillary antrostomy, and we saw a sponge-like tumor. The AP study showed partialy immature bone tissue, and within its trabeculae there was fibrous connective tissue, with no evidence of significant atypia, leading us to consider it a cement-ossifying fibroma. We proposed surgical excision, but the patient refused it.
DISCUSSION
Stesioneuroblastoma
The stesioneuroblastoma is a neurogenic tumor that originates from the olfactory region neuroepithelium3. It corresponds to 6% of the malignant nasal and paranasal tumors; and to 0.3% of the upper airway and digestive tract cancers4. It happens at any age, with similar prevalence in both genders5. It was initially described by Berger et al. in 1924, under the term "esthésioneuroépithéliome olfactif"5. Between 1924 and 1989, less than 300 cases were reported5. This figure almost reached 1,000 after a literature review carried out by Broich et al., and most of them were diagnosed after 1997, probably because of histological diagnosis difficulty in small-cell tumors and restricted access to immunohistochemistry5.
Symptoms are unspecific and common to most tumors and benign diseases of the nasal cavity - unilateral nasal obstruction and epistaxis are the most frequent ones5. Headache, visual disorders and hyposmia are also reported3,5. Kadish's classification divides the tumors in three stages: A (tumors restricted to the nasal cavity), B (tumors involving the nasal cavity and paranasal sinuses) and C (tumors extending to beyond the paranasal sinuses -orbit, skull base or metastasis)5.
CT scan is the radiologic exam of choice, which shows a homogeneous mass in the nasal cavity, of soft tissue density and uniform and moderate contrast uptake6. It is important to asses a possible erosion of the lamina papyracea, lamina cribosa and ethmoidal fovea. MRI is useful to determine intraorbital or intracranial tumor extension6.
The treatment proposed for the case is the one advocated by most authors: cranio-facial resection followed by radiotherapy3,6,7. A metanalysis carried out in 2001 correlated the survival rate in five years with treatment modality - 65% with surgery and radiotherapy, 51% with radiotherapy and chemotherapy, 48% surgery alone, 47% with surgery plus radiotherapy and chemotherapy and 37% with radiotherapy alone7.
Neck lymphadenopathy is an important prognostic factor, and the survival rate is 64% in patients without neck metastasis and 29% in patients with metastasis7. Local recurrence happens in 15 to 20% of the cases6, which has been reported even after 10 years of initial treatment, requiring prolongued treatment, especially in young patients. Distant metastasis after local lesion control is relatively rare (4 to 8%)5.
Giant Cells Central Granuloma (GCCG)
GCCG was only recognized as a clinical entity after 1953, and was first described by Jaffe who called it giant cells central "repair" granuloma9. It is a rare pathology, corresponding to less than 7% of the benign maxilla and mandible tumors10. More than 70% of the cases affect the mandible, anteriorly to the 1st molar9, and the mandible/maxilla ratio is of 2-3:1. There are reports of GCCG, however, in the temporal bone, paranasal sinuses, skull base and orbit, among others10. More than 60% of the cases happen in patients below 30 years of age, with predominance for females11.
The pathogenesis of GCCGs is still little understood, and there are controversies regarding its neoplastic origin or reactional12. Recent studies show proliferative GCCG activity, suggesting a deregulation in the cell cycle that can contribute to the disease's pathogeny. Alterations in the expression of cyclin D1, a protein that regulates the cell cycle, were found in GCCGs and in giant cell tumors (GCT)12. Similar immunohistochemistry patterns for other proteins that regulate the cell cycle (MDM2 and p53) were also observed in GCTs and GCCGs12. Having this in mind, some authors consider these diseases a continuation of the same disease12,13.
According to clinical behavior, the GCCGs are classified in aggressive and non-aggressive forms11. The former has a slow growth, usually asymptomatic, with no invasion of the bone cortical and low recurrence likelihood11. Now, the aggressive form has fast and painful growth, with cortical invasion and tendency to recurr11. No histological or immunohistochemical characteristic has proven to have any correlation with tumor behavior11,14.
Histologically we see CD68+ multinucleated giant cells and mononuclear cells in a fibrous stroma12,14. The GCCG does not have pathognomonic radiologic alterations, and it can simulate other bony lesions10. The majority (87.5%) present as a radiolucent expansive lesion, uni or multilocular, with well outlined borders (56%) or not so well defined borders (30%)11. More aggressive maxilla lesions may invade the maxillary sinus floor, the orbit and the nasal cavity11. One may find mandible cortical bone rupture, teeth shifting or tooth root resorption11.
Treatment is based on en-bloc surgical resection or resection by curettage, depending on the clinical and radiologic aspect of the lesion9,11. Since it is a more aggressive lesion, we chose en-bloc resection in this case, and we attained a very satisfactory result. One study showed a 50% recurrence rate after curettage, compared to only 10% after complete lesion resection10. The most used alternative treatment modalities are: steroid injection; calcitonin injection or intranasal spray; and alpha-2a interferon injection9.
Extramedullary Plasmocytoma
The plasmocytoma is a rare neoplasia originated from B cells. It may present as three variants: multiple myeloma (MM), osseous solitary plasmocytoma and extramedullary plasmocytoma (EMP)15,16. MM, initially described in 184616, is the most common and spread form of plasmatic cells tumor. The osseous solitary plasmocytoma is characterized by trabeculated and multicystic intra-osseous lesions, usually involving the spine, pelvis and femur16.
EMPs represent about 4% of the non-epithelial nasosinusal tumors and about 0.4% of the head and neck tumors16. It develops in the submucosal tissues of the upper airways in 80% of the cases, with decreasing incidence in the nose, paranasal sinuses, nasopharynx, palatine tonsil, thyroid, gums and hypopharynx15. It prevails in males at the rate of 3-4:1, and it is more frequent in those patients above 40 years of age17.
The most common symptoms of nasal cavity EMP, as seen in our case, are progressive nasal obstruction and intermittent epistaxis15,17. Pain is uncommon, unless there is secondary infection or bone destruction16. Physical or endoscopic exams of the nasal cavity reveal a submucosal, polypoid and lobulated tumor18, usually not ulcerated and brittle15. The anterior rhinoscopy of this case did not show tumoral lesion, calling for the need of complementary diagnostic approaches.
The CT scan shows an expansive mass, usually homogeneous, with minimum to no bone erosion. Light microscopy shows groups of plasmocytic cells with different degrees of differentiation, varying from mature and small cells to immature and multinucleated16, associated to a rearrangement of adjacent structures.
Laboratorial exams must be ordered in order to rule out MM: CBC, protein electrophoresis, bone marrow aspirate and biopsy, electrolytes, radiological studies of the skeleton, among others. Some patients, between 5% and 32%, will develop systemic manifestations of MM after some years, which require prolongued follow up19. Treatment is based on surgery and complementary radiotherapy. EMPs are radiosensitive and regional control can be achieved in more than 80% of the cases17,19.
Nasosinusal Hemangiopericytoma (NSHP)
The NSHP is a rare mesenchymal tumor of the nasosinusal region, characterized by a pattern of prominent perivascular growth20. Since its initial description in 1942, the definition of this disease as a specific clinical entity has been questioned21. Despite the terminology, it is believed that this disease represents a neoplasia that is clinically and pathologically different from the conventional soft tissue hemangiopericytoma (STHP)20,21. Some authors associate it with the glomus tumor, considering it a variant of the former or even a hybrid between the glomus tumor and the STHP20, due to the similarities in biological behavior, cytological profile and immunophenotypes.
Between 15% and 30% of all STHP happen in the neck and head region, they account for less than 1% of vascular neoplasias21,22. The NSHP affects preferentially those patients in the sixth and seventh decades of life in both genders20,21. The most common clinical picture is unilateral nasal obstruction and/or epistaxis, with a polypoid mass in the nasal cavity20,21. Radiologic exams reveal opacification caused by polypoid mass, sometimes associated to bone erosion and sclerosis 21.
Histologically, the NSHP is a submucosal, non-capsulated lesion, with diffuse growth pattern and prominent perivascular hyalinization20,21. It has a myoid appearance without cellular pleomorphism, with tumoral cells organized in sheets or fascicles20,21. They bear no specific immunohistochemical marker. There is frequent expression of vimentin, actin and smooth muscle actin20,21. Contrary to the STHP, it does not manifest CD3420,21. In the case hereby presented, the tumoral cells manifested only vimentin.
In general, NSHPs are very resistant. They manifest aggressive behavior as in the case hereby presented, they are uncommon and there are reports in the literature of lesions with local destructive potential and metastasis21. Histological characteristics that may be associated with aggressive behavior include a larger number of mitotic figures, increase in cellularity with cell pleomorphism and necrosis21. Large lesions (larger than or equal to 6.5cm) and incomplete first resection are all associated with a worse prognosis21.
Treatment is surgical, varying from simple polypectomy to resection with broad free margins20,21. Local recurrence happens in 17% to 40%21 of the cases, probably due to residual tumor. Prognosis is favorable, with 5-year-disease-free survival rate of 74.2% and in 10 years it is 64.4%21. Radio and chemotherapy can be used for metastatic disease and residual or non-resectable primary tumors21.
Neurofibroma
Benign neural tumors, represent about 45% of the head and neck tumors23. The neurofibroma, a neoplasia originated from the Schwann cells, is the most common type, affecting most frequently the skin23. About 25% to 45% of neurofibromas happen in the head and neck, and only 4% involve the nasal cavity and paranasal sinuses24.
It manifests as isolate lesion or multiple tumors, as it happens to the Type 1 neurofibromatosis (NF1) or Von Recklinghausen's disease23. Almost all patients with NF1 will develop a neurofibroma at some point in their lives25. The potential for malignization is of 2.6%, and in the syndrome it varies between 3% and 15%24.
Despite being non-capsulated, these are well-outlined lesions. Light microscopy shows a proliferation of all the elements of the peripheral nerve, spread in a loose and disorganized pattern in a myxoid stroma. There is a predominance of Schwann cells with fusiform nuclei23. This disarranged architecture helps differentiate it from schwannomas. Treatment is surgical24,25.
Cement-Ossifying Fibroma - COF
The COF is a rare, non-odontogenic mesodermic tumor26,27. According to the classification from the World Health Organization, it is considered a variant of the cementifying fibroma, which represents a subgroup of cementomas27. The cementomas, fibrous-osseous lesion with cement, are divided in four subgroups, according to their clinical, histopathological and radiological characteristics: benign cementoma, cementifying fibroma, cement periapical dysplasia and gigantiform cementoma26,28.
With still controversial etiopathogenesis, it is believed that these lesions stem from the periodontal membrane, which contains blast cells capable of forming bone, fibrous tissue and cement26. Tooth extraction or traumas have been considered possible causal factors26.
The majority occur in the molar and pre-molar regions of the mandible29. They may also involve the maxilla and, more rarely, other paranasal sinuses. Some studies show their predilection for females, more frequent in the 4th and 5th decades of life29. They usually manifest as an asymptomatic mass, well outlined and slow growth, diagnosed in a routine radiologic exam29. It may be associated with local pain, teeth loss or dental occlusion alterations29. Large maxillary lesions may cause facial asymmetry, rhinorrhea, proptosis and diplopia29. There are reports of aggressive tumors, especially when located in the maxilla and ethmoid26.
Radiologically, they manifest as a well outlined mass, with soft tissue density and foci of ossification, there may be bone destruction30. Light microscopy shows fibrous tissue with spheroid bony lamellar trabeculae, permeating a fibrous stroma in close association with fibroblasts. Masses of acellular basophile material are shown as concentric lines, identified as cement30.
Treatment is surgical, and there are reports of remission after incomplete resection26,29. In principle, radiotherapy is contra-indicated, because of tumor resistance and the possibility for malignization29,30.
CONCLUSION
Non-epithelial tumors of the nasal cavity and paranasal sinuses are rare pathologies, with extremely varied etiopathology, clinical behavior, treatment and prognosis. The lack of symptom specificity usually delays diagnosis. Having all of this in mind, the physician must be attentive to the diverse number of diseases that may affect this region, and histopathology is extremely necessary for properly approaching these cases.
REFERENCES
1. Cahali BM, Sennes LU, Murakami MS, D'Antonio WE, Ikino CM, Santoro PP et al. Tumores nasossinusais malignos: espectro de apresentação nos últimos 15 anos. Rev Bras Otorrinolaringol 1999;65(2):296-300.
2. Valle A. Epidemiologia do câncer de cabeça e pescoço no Brasil e no mundo. Em: Tratado de otorrinolaringologia. 1ª ed. São Paulo: Roca; 2002. 1: 860.
3. Theilgaard SA, Buchwald C, Ingeholm P, Larsen SK, Eriksen JG, Hansen SH. Esthesioneuroblastoma: a Danish demographic study of 40 patients registered between 1978 and 2000. Acta Otolaryngol 2003;123(3):433-9.
4. Svane-Knudsen, Jorgensen KE, Hansen O, Lindgren, Marker P. Cancer of the nasal cavity and paranasal sinuses: a series of 115 patients. Rhinology 1998;36(1):12-4.
5. Lund VJ, Howard D, Wei W, Spittle M. Olfactory neuroblastoma: past, present and future? Laryngoscope 2003;113(3):502-7.
6. Dulguerov P, Calcaterra T. Esthesioneuroblastoma: the UCLA experience 1970-1990. Laryngoscope 1992;102(8):843-9.
7. Dulguerov P, Allal AS, Calcaterra TC. Esthesioneuroblastoma: a meta-analysis and review. Lancet Oncol 2001;2:683-90.
8. Ferlito A, Rinaldo A, Rhys-Evans P. Contemporary clinical commentary: esthesioneuroblastoma: an update on management of the neck. Laryngoscope 2003;113(11):1935-8.
9. Oda D. Alternative treatment for central giant cell "reparative" granuloma. Advancer in Anatomic Pathology 2003;10(2):110.
10. Plontke SK, Adler C, Gawlowski J et al. Recurrent giant cell reparative granuloma of the skull base and the paranasal sinuses presenting with acute one-sided blindness. Skull Base 2002;12(1):9-16.
11. Carlos R, Sedano HO. Intralesional corticosteroids as an alternative treatment for central giant cell granuloma. Oral Surg Oral Med Oral Pathol 2002;93(2):161-6.
12. Kauzman A, Li S, Bradley G et al. Central giant cell granuloma of the jaws: assessment of cell cycle proteins.J Oral Pathol 2004;33(3):170-6.
13. Kauzman A, Li S, Bradley G et al. Cell cycle alterations in central giant cell granuloma of the jaws: a comparison with giant cell tumor of bone. Oral Surg Oral Med Oral Pathol 2003;96(3):299.
14. Regezi JA. Odontogenic Cysts, Odontogenic Tumors, Fibroosseous, and Giant Cell Lesions of the Jaws. Modern pathol 2002;15(3):331-41.
15. Fernandes AM, Podovani JA, Maníglia JP. Plasmocitoma extramedular de nasofaringe: Relato de um caso e revisão da literatura. Rev Bras Otorrinolaringol 1998;64(3):296-8.
16. Arrarte J, Barra M, Fontoura S, Peduzzi F, Junior H. plasmocitoma extra medular de nariz, relato de un caso. An ORL Mex 2000;45(3):118-20.
17. Sahin S, Dulundu H, Coskun H. Nasal plasmocytoma, proceedings of the XV world congress of ORL, head and neck surgery 1993;2:486-88.
18. Robbins SL, Cotran RR, Kumar VK, Collins T. Robbins pathologic basis of disease. 6a ed. Philadelphia: Saunders 1999;763-4.
19. Courtmans I, Pigeolet Y, Hedayat A, Vilain J. Upper airways locations of plasmocytoma. Acta Oto-Rhino-Laryngol Belgica 2000;54(4):487-90.
20. Li X, Hisaoka M, Morio T, Hashimoto H. Intranasal pericytic tumors (glomus tumor and sinonasal hemangiopericytoma-like tumor): Report of two cases with review of the literature. Patho international 2003;53(5):303-8.
21. Thompson L, Miettinen M, Wenig B. Sinonasal-Type Hemangiopericytoma: A Clinicopathologic and Immunophenotypic Analysis of 104 Cases Showing Perivascular Myoid Differentiation. Am J of Surg Patho 2003;27(6):737-49.
22. Sabini P, Josephson G, Yung R, Dolitsky J. Hemangiopericytoma presenting as a congenital midline mass. Arch Oto Head and Neck Surg 1998;124(2):202-4.
23. Souza L, Oliveira J, Freitas T, Carvalho R. Neurofibroma paciniano: relato de um caso raro de localização intra-oral. Rev Bras Oto 2003;69(6):851-4.
24. Silva V, Agra L, Freitas M, Pinheiro S. Neurofibroma nasal: relato de caso e revisão de literatura. Rev Bras Otorrinolaringol 1999;65(2):172-4.
25. Packer R, Gutmann D, Rubenstein A et al. Plexiform neurofibromas in NF1: Toward biologic-based therapy. Neurology 2002;58(10):1461-70.
26. Jacobs J, Berg H. Destructive cemento-ossifying fibroma of the maxilla. Ear Nose Throat J 1999;69(12):805-8.
27. Bowyer J, Majid M, Ah-Fat F et al. Giant cemento-ossifying fibroma of the maxilla causing proptosis in a young patient. J Pediatr Ophthalmol Strazismus 2001;38(6):359-62.
28. Selmani Z, Anttila J, Mertakorpi J et al. Cemento-ossifying fibroma of the etmoidal sinus in a child presenting with isolated pain in the nasal region. J Craniofacial Surg 2004;15(2):215-7.
29. Sciubba J, Goldstein M, Cohen A, Abramson A. A destructive maxillary cemento-ossifying fibroma following maxillofacial trauma. Laryngoscope 1984;94(6):810-50.
30. Fujimoto Y, Katho M, Miyata M et al. Cystic cemento-ossifying fibroma of the etmoidal cells. J Laryngol Otol 1987;101(9):946-52.
1 ENT Resident - HGF SESA/SUS.
2 ENT Residency Program Preceptor - Hospital Geral de Fortaleza, Otorhinolaryngologist.
3 MD. Pathologist. Assistant Professor of General Pathology - Federal University of Ceará Medical School.
4 ENT Resident - HGF SESA/SUS.
5 ENT Resident - HGF SESA/SUS.
Hospital Geral de Fortaleza - SESA/SUS.
Send correspondence to: Rua Fausto Cabral 95 apto. 701 Bairro Vicente Pinzon 60175-415 Fortaleza CE.
Paper submitted to the ABORL-CCF SGP (Management Publications System) on March 31th, 2005 and accepted for publication on May 24th, 2005. cod. 204.
Print: ![]()