

Ano: 2008 Vol. 74 Ed. 2 - Março - Abril - (25º)
Seção: Relato de Caso
Páginas: 307 a 314
 PDF PT
PDF PT Tumores nasossinusais raros: série de casos e revisão de literatura
Rare nasosinusal tumors: Case series and literature review
Autor(es): Roberta de Paula Araújo 1, Érika Ferreira Gomes 2, Dalgimar Beserra de Menezes 3, Lidiane Maria de Brito Macedo Ferreira 4, Adson Sales do Nascimento Rios 5
Palavras-chave: cavidade nasal, neoplasia
Keywords: nasal cavity, neoplasm
Resumo:
Os tumores nasossinusais são patologias pouco freqüentes na prática clínica. Aproximadamente 0,8% de todos os cânceres humanos localizam-se nessa região. Apesar de rara, a neoplasia nasossinusal manifesta-se habitualmente através de sintomas inespecíficos e comuns a inúmeras patologias inflamatórias. Este estudo se propõe a descrever uma série de casos de tumores nasossinusais não-epiteliais raros, incluindo estesioneuroblastoma, granuloma central de células gigantes, plasmocitoma extramedular, hemangiopericitoma sinonasal, neurofibroma e fibroma cemento-ossificante, diagnosticados no Hospital Geral de Fortaleza, SESA/SUS. Faz-se uma breve revisão de literatura de cada patologia, salientando-se a necessidade do diagnóstico anatomopatológico preciso para condução adequada de cada caso.
Abstract:
Tumors of the nasal cavity and paranasal sinuses are unusual pathologies found in clinical practice. Approximately 0.8% of all human cancers are located in this area. Despite being rare, nasosinusal neoplasms usually manifest through nonspecific symptoms that are common to numerous inflammatory pathologies. The aim of this study is to describe a series of rare nasosinusal tumors, including esthesioneuroblastomas, central giant cell granulomas, extramedullary plasmocytomas, nasosinusal hemangiopericytomas, neurofibromas and cemento-ossifying fibromas, diagnosed at the Fortaleza General Hospital. We, hereby, briefly review each of the aforementioned pathologies, stressing the need for a precise histological diagnosis for proper treatment in each case.
![]()
INTRODUÇÃO
Os tumores malignos nasossinusais são raros, correspondendo a menos de 3% dos cânceres de cabeça e pescoço e a 0,8% de todos os cânceres humanos1. Aproximadamente 55% originam-se dos seios maxilares, 35% da cavidade nasal, 9% dos seios etmoidais e 1% dos seios frontais e esfenoidais2. Nos EUA, a incidência de tumores de cavidade nasal é de menos de 1 caso em 100 mil pessoas por ano2. Com exceção de tumores não-epiteliais, o câncer nasossinusal é uma patologia de adultos, sendo mais freqüente em homens após a 5a década de vida2.
Os tumores dessa região apresentam-se geralmente com sintomas inespecíficos e comuns, inclusive, a patologias inflamatórias. Obstrução nasal (61,2%), epistaxe (40,8%), algia facial (39,2%) e infecção local (23,9%) são as queixas iniciais mais relatadas1. Esse fato, aliado à baixa incidência dessas lesões e, muitas vezes, à dificuldade do diagnóstico histopatológico, contribuem para o retardo do diagnóstico e do tratamento na maioria dos casos.
APRESENTAÇÃO DE CASOS CLÍNICOS
Caso 1
ATS, sexo feminino, 16 anos, procedente de Acarape/CE, procurou o serviço com queixa de epistaxe seguida de obstrução nasal à direita há cerca 3 anos. À rinoscopia anterior, visualizava-se lesão tumoral hiperemiada, friável, ocupando a porção superior da fossa nasal direita (FND), estendendo-se até o meato médio.
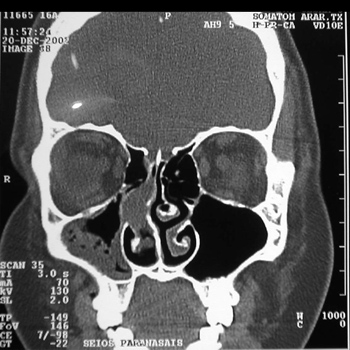
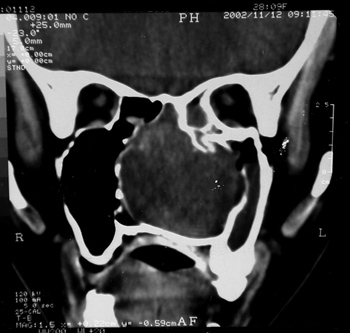
Tomografia computadorizada (TC) de cavidade nasal e seios paranasais mostrava formação densa de aspecto polipóide, bem definida, em FND, obliterando meato médio e recesso esfenoetmoidal, com perda de definição de concha média e assoalho das células aéreas etmoidais posteriores ipsilaterais. Evidenciava-se também espessamento mucoso em seio maxilar e esfenoidal direitos (Figura 1).
Figura 1. TC corte coronal de Estesioneuroblastoma (caso 1).
Biópsia incisional seguida de tamponamento nasal foi realizada ambulatorialmente. O estudo anatomopatológico (AP) revelou aglomerados de pequenas células de núcleos hipercromáticos redondos em meio a estroma fibrilar, sendo o quadro compatível com estesioneuroblastoma. Diante do diagnóstico da lesão, classificada como Kadish B (envolvimento da cavidade nasal e seios paranasais), foi proposta ressecção via degloving médio-facial, seguida de radioterapia (RxT). Ainda no pré-operatório, a paciente apresentou epistaxe volumosa, incitando a realização de arteriografia e embolização do tumor por cateterização seletiva de artéria maxilar direita.
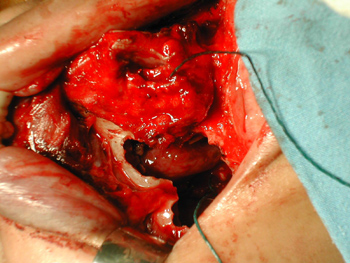
No oitavo dia pós-embolização, foi realizada ressecção crânio-facial do tumor. A lesão ocupava porção superior da FND, medindo aproximadamente 4cmx3cmx1,5cm. Sinusiotomia maxilar anterior mostrou ausência de tecido tumoral em seio maxilar direito (Figura 2). Parede medial do seio maxilar foi ressecada e lâmina crivosa do etmóide foi brocada até a dura-máter para garantir margem cirúrgica. A paciente foi então encaminhada para RxT, estando atualmente no 8o mês de seguimento, sem evidência de recidiva.
Figura 2. Degloving médio-facial com sinusiotomia maxilar anterior de Estesioneuroblastoma (caso 1).
Caso 2
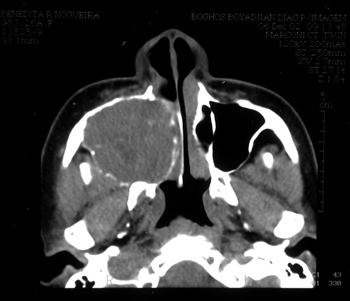
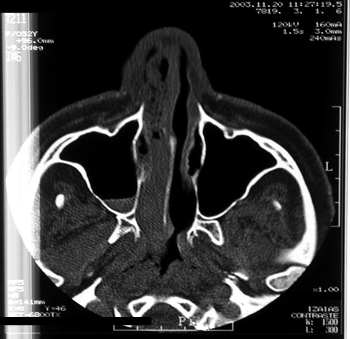
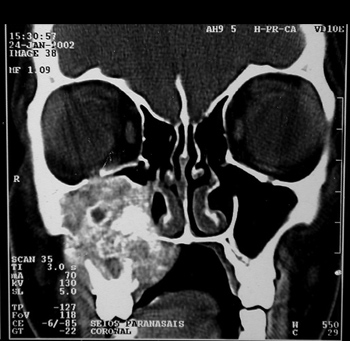
BRN, sexo feminino, 26 anos, procedente de Fortaleza/CE, procurou o serviço com queixa de abaulamento de hemiface e obstrução nasal à direita, com cerca de 8 meses de evolução. Relatava excisão dentária (1o pré-molar superior direito) há cerca de 1 ano. TC mostrou volumosa formação sólida expansiva, heterogênea, ocupando seio maxilar direito, com expansão e pequenas áreas de invasão da cortical. A lesão estendia-se para palato duro e processos alveolares dentários ipsilaterais, para FND e obliterava parcialmente a fissura orbitária inferior (Figura 3).
Figura 3. TC corte axial de GCCG (caso 2).
Ao realizar-se biópsia via antrostomia sublabial, visualizou-se ausência de parede óssea anterior do seio maxilar D e tumor encapsulado e friável, de aspecto carnoso e vascularizado. Estudo AP revelou secções de tumor que reproduz células gigantes multinucleadas, inseridas em estroma fibroso de densidade variável, com proliferação de células ovóides e fusiformes, em quadro compatível com granuloma central de células gigantes (GCCG). O tratamento proposto foi maxilarectomia direita via degloving médio-facial. A paciente encontra-se no 24o mês de acompanhamento, sem evidência de recidiva tumoral (Figura 4).
Figura 4. TC corte coronal de pós-operatório de GCCG (caso 2).
Caso 3
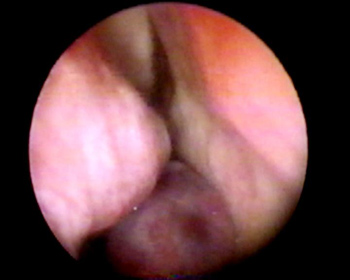
CAS, sexo feminino, 52 anos, procedente de Fortaleza/CE, queixava-se de epistaxe há cerca de 3 anos seguida de obstrução nasal à direita. O exame físico otorrinolaringológico era normal. À nasofibroscopia, visualizava-se tumoração em assoalho da FND, com extensão para nasofaringe (Figura 5). TC de seios da face mostrava lesão expansiva homogênea, com densidade de partes moles em FND e nasofaringe, sem evidência de erosão óssea (Figura 6). Biópsia incisional foi realizada, demonstrando, à microscopia, neoformação vascular e conjuntiva com intensa infiltração de células plasmocitóides e revestimento do tipo respiratório, compatível com granuloma plasmocitário. Exames laboratoriais foram solicitados no intuito de descartar doença sistêmica (mieloma múltiplo). Exames hematológicos, aspirado e biópsia de medula óssea e eletroforese de proteínas estavam dentro dos padrões de normalidade. Excisão cirúrgica da lesão foi realizada por via endoscópica, sem evidências de invasão óssea ou de estruturas adjacentes. A paciente foi encaminhada para RxT complementar, estando no 8o mês de seguimento, sem sinais de recidiva tumoral ou de doença sistêmica.
Figura 5. Nasofibroscopia de Plasmocitoma Extramedular FND (caso 3).
Figura 6. TC corte axial de Plasmocitoma Extramedular (caso 3).
Caso 4
LDS, 15 anos, sexo feminino, procedente de Fortaleza/CE, procurou o serviço com história de odontalgia e lesão bolhosa em palato à esquerda, seguidas de epistaxe, abaulamento de hemiface e exoftalmia ipsilaterais com aproximadamente um ano de evolução. TC de seios da face demonstrava lesão expansiva em FNE, com extensão aos ápices orbitários anteriormente e região supra-selar, e com destruição de lâminas pterigóides. A lesão estendia-se para seio esfenoidal, maxilar e palato duro ipsilaterais, sendo hipocaptante (Figura 7).
Figura 7. TC corte coronal de Hemangiopericitoma Sinonasal (caso 4).
À primeira tentativa de biópsia, houve sangramento importante, sendo então realizada arteriografia e embolização do tumor, que apresentou regressão expressiva após o procedimento. Realizou-se biópsia excisional via antrostomia maxilar anterior, sendo visualizada apenas cápsula tumoral e tecido necrótico em volumosa cavidade no seio maxilar.
Estudo AP mostrou tecido conjuntivo com infiltrados mistos de células inflamatórias e fendas vasculares com colarete de células de núcleos redondos; áreas de deposição de material mucinoso e depósito de pigmento hemossiderínico. Concluiu-se corresponder a uma lesão angiomatóide sem sinais de malignidade, com diagnóstico diferencial entre glomangioma e hemangiopericitoma sinonasal. Células tumorais dispostas em torno de espaços vasculares expressavam apenas vimentina à imunohistoquímica, sendo negativas para actina, actina de músculo liso, CD31 e CD34, sendo mais compatível com hemangiopericitoma sinonasal.
Três meses após intervenção cirúrgica, a paciente retornou ao serviço com queixa de epistaxe. À nasofibroscopia, foi visualizado tumor residual em assoalho de FNE com extensão para palato mole. TC demonstrava lesão que media aproximadamente 5cm X 3cm, com destruição e rarefação óssea, envolvendo assoalho da órbita e parede lateral e assoalho do seio maxilar. A lesão destruía clívus, etmóide e osso esfenóide à esquerda, com invasão da região para-selar e envolvimento do seio cavernoso (Figura 8).
Figura 8. TC corte axial de Hemangiopericitoma sinonasal recidivado (caso 4).
A paciente foi submetida à RxT e quimioterapia (QT), evoluindo para óbito cerca de um ano após o diagnóstico, com quadro clínico compatível com hemorragia intracraniana.
Caso 5
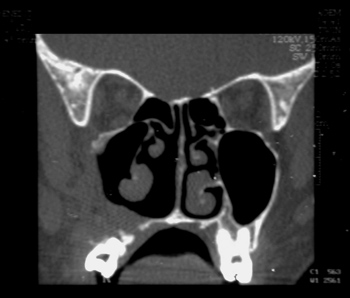
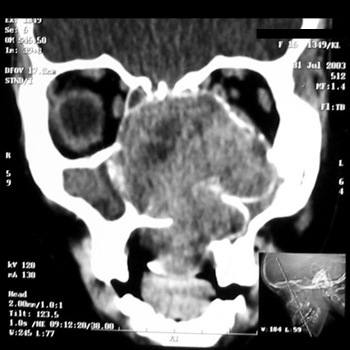
AAGL, sexo feminino, 23 anos, procedente de Maranguape/Ce, com história de epistaxe e obstrução nasal à esquerda há cerca de 5 anos. Apresentava, ao exame físico, tumoração hiperemiada e não-sangrante em FNE, com abaulamento de pirâmide nasal, de região maxilar e de palato ipsilaterais. TC mostrava lesão expansiva em FNE, ocupando rinofaringe, seios maxilar e etmoidal ipsilaterais, com desvio de septo nasal para direita e sem evidência de lise óssea (Figura 9).
Figura 9. TC corte coronal de Neurofibroma (caso 5).
Biópsia incisional foi realizada, revelando, à microscopia, proliferação de células alongadas e fusiformes, às vezes com núcleos arregimentados, arranjadas em feixes, compatível com neurofibroma. Antrostomia maxilar sublabial foi realizada para ressecção da lesão, que media aproximadamente 9cmx9cmx2cm. Há 17 meses em seguimento pós-operatório, não há evidência de recidiva tumoral.
Caso 6
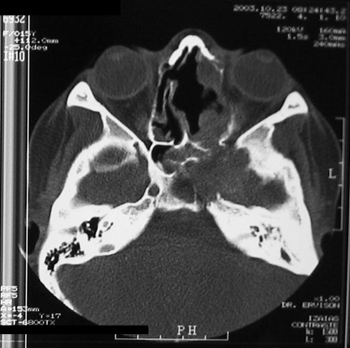
MVPM, sexo feminino, 15 anos, apresentava abaulamento de região infraorbitária à direita, ulceração periodontal de molares superiores e abaulamento submucoso de palato duro ipsilaterais com cerca de 1 ano de evolução. Negava epistaxe ou obstrução nasal. TC demonstrava lesão expansiva com calcificações concêntricas em seio maxilar direito, com destruição de suas paredes e invasão de palato duro (Figura 10).
Figura 10. TC corte coronal de fibroma cemento-ossificante (caso 6).
Foi realizada biópsia incisional via antrostomia maxilar anterior, visualizando-se tumor de aspecto de osso esponjoso. Estudo AP revelou tecido ósseo, em parte imaturo, entre cujas trabéculas havia tecido conjuntivo fibroso, sem evidência de atipia significativa, em quadro compatível com fibroma cemento-ossificante. Foi proposta ressecção cirúrgica da lesão, sendo que a paciente recusou o procedimento.
DISCUSSÃO
Estesioneuroblastoma
Estesioneuroblastoma é um tumor neurogênico, originado do neuroepitélio da região olfatória3. Corresponde a 6% dos tumores nasais e paranasais malignos e a 0,3% dos cânceres do trato aerodigestivo superior4. Ocorre em qualquer idade, com prevalência semelhante em ambos os sexos5. Foi inicialmente descrito por Berger et al. em 1924, sob o termo "esthésioneuroépithéliome olfactif"5. Entre 1924 e 1989, menos de 300 casos haviam sido relatados5. Esse número chegou a quase 1000 após revisão de literatura realizada por Broich et al., sendo a maioria diagnosticada a partir de 1997, provavelmente devido à dificuldade do diagnóstico histológico em tumores de pequenas células e ao acesso até então restrito à imunohistoquímica5.
Os sintomas são inespecíficos e comuns à maioria dos tumores e a doenças benignas da cavidade nasal, sendo obstrução nasal unilateral e epistaxe os mais freqüentes5. Cefaléia, distúrbios visuais e hiposmia também são relatados3,5. A classificação de Kadish divide os tumores em 3 estágios: A (tumores confinados à cavidade nasal), B (com envolvimento da cavidade nasal e seios paranasais) e C (com extensão para além dos seios paranasais - órbita, base de crânio ou metástase)5.
TC é o exame radiológico de escolha, mostrando massa homogênea em fossa nasal superior, com densidade de tecidos moles e captação uniforme e moderada de contraste6. Deve-se avaliar possível erosão de lâmina papirácea, lâmina crivosa e fóvea etmoidal. Ressonância nuclear magnética é útil para determinar extensão intraorbitária ou intracraniana6.
O tratamento proposto para o caso é o defendido pela maioria dos autores: ressecção crânio-facial seguida de RxT3,6,7. Meta-análise realizada em 2001 correlacionou a taxa de sobrevida em 5 anos com a modalidade de tratamento, sendo de 65% com cirurgia e RxT, 51% com RxT e QT, 48% só com cirurgia, 47% com cirurgia associada a RxT e QT e 37% só com RxT7.
Linfadenopatia cervical é um fator prognóstico importante, sendo a taxa de sobrevivência de 64% em pacientes sem metástase cervical e de 29% em pacientes com metástase7. Recorrência local ocorre em 15 a 20% dos casos6, tendo sido relatada mesmo após 10 anos do tratamento inicial, o que exige acompanhamento prolongado, especialmente em pacientes jovens. Metástase à distância após controle regional da lesão é relativamente rara (4 a 8%)5.
Granuloma Central de Células Gigantes (GCCG)
O GCCG foi reconhecido como entidade clínica a partir de 1953, sendo descrito pela primeira vez por Jaffe sob o termo granuloma central "reparador" de células gigantes9. É uma patologia rara, correspondendo a menos de 7% dos tumores benignos de maxila e mandíbula10. Mais de 70% dos casos ocorrem na mandíbula, anteriormente ao 1o molar9, sendo a relação mandíbula/ maxila de 2-3:1. Há relatos de GCCG, entretanto, no osso temporal, seios paranasais, base de crânio e órbita, entre outros10. Mais de 60% dos casos ocorrem em pacientes menores de 30 anos, com predominância pelo sexo feminino11.
A patogênese do GCCG é ainda pouco compreendida, havendo controvérsias quanto à origem neoplásica ou reacional da lesão12. Estudos recentes demonstraram atividade proliferativa no GCCG, sugerindo que uma desregulação no ciclo celular pode contribuir para a patogênese da lesão. Alterações na expressão de ciclina D1, uma proteína reguladora do ciclo celular, foram encontradas em GCCGs e em tumores de células gigantes (TCG)12. Padrões semelhantes de imunohistoquímica para outras proteínas reguladoras de ciclo celular (MDM2 e p53) foram também observados em TCG e GCCG12. Em vista disso, alguns autores consideram essas patologias um continuum da mesma doença12,13.
De acordo com o comportamento clínico, classificam-se os GCCGs em formas não-agressiva e agressiva11. A primeira apresenta crescimento lento, geralmente assintomático, sem invasão da cortical do osso e com baixa probabilidade de recorrência11. Já a forma agressiva, apresenta crescimento rápido, doloroso, com invasão de cortical e alta tendência a recorrer11. Nenhuma característica histológica ou imunohistoquímica tem demonstrado correlação com o comportamento do tumor11,14.
Histologicamente, evidenciam-se células gigantes multinucleadas CD68+ e células mononucleares em um estroma fibroso12,14. GCCG não exibe alterações radiológicas patognomônicas, podendo simular outras lesões ósseas10. A maioria (87,5%) apresenta-se como lesão expansiva radiolucente, uni ou multilocular, com bordas bem definidas (56%) ou pouco definidas (30%)11. Lesões de maxila mais agressivas podem invadir assoalho do seio maxilar, órbita e cavidade nasal11. Pode-se encontrar ruptura da cortical da mandíbula, deslocamento de dentes ou reabsorção da raiz11.
O tratamento consiste na ressecção cirúrgica em bloco ou por curetagem, dependendo da apresentação clínica e radiológica da lesão9,11. Por tratar-se de uma lesão mais agressiva, optou-se pela ressecção em bloco no caso apresentado, obtendo-se resultado satisfatório. Um estudo demonstrou 50% de recorrência após curetagem comparado com somente 10% após ressecção completa da lesão10. Os tratamentos alternativos mais usados são: injeção de esteróides; injeção ou spray nasal de calcitonina; e injeção de interferon alfa-2a9.
Plasmocitoma Extramedular
Plasmocitoma é uma neoplasia rara originada de linfócitos B. Pode apresentar-se como três variantes: mieloma múltiplo (MM), plasmocitoma solitário ósseo e plasmocitoma extramedular (PEM)15,16. O MM, inicialmente descrito em 184616, é a forma mais comum e disseminada de tumor de células plasmáticas. O plasmocitoma solitário ósseo, por sua vez, caracteriza-se por lesões intra-ósseas trabeculadas e multicísticas, acometendo com maior freqüência a coluna, pelve e fêmur16.
PEM representa cerca de 4% dos tumores nasossinusais não epiteliais e cerca de 0,4% dos tumores de cabeça e pescoço16. Desenvolve-se em tecido submucoso de vias aéreas superiores em 80% dos casos, com incidência decrescente em nariz, seios paranasais, nasofaringe, tonsila palatina, tireóide, gengiva e hipofaringe15. Predomina no sexo masculino na proporção de 3-4:1, sendo mais freqüente após os 40 anos de idade17.
Os sintomas mais comuns do PEM de cavidade nasal, como exemplificado no caso, são obstrução nasal progressiva e epistaxe intermitente15,17. Dor é incomum, a menos que haja infecção secundária ou destruição óssea16. O exame físico ou endoscópico da cavidade nasal revela tumoração submucosa, polipóide, de aspecto lobulado18, geralmente não ulcerada e friável15. À rinoscopia anterior do caso apresentado, não se visualizava lesão tumoral, alertando à necessidade de uma avaliação diagnóstica complementar.
TC demonstra massa expansiva, geralmente homogênea, com erosão óssea mínima ou ausente. À microscopia, evidenciam-se grupos de células plasmocitárias com graus de diferenciação variando de células maduras e pequenas a imaturas e multinucleadas16, associadas a rearranjo de estruturas adjacentes.
Exames laboratoriais devem ser solicitados no intuito de excluir MM, como hemograma, eletroforese de proteínas, aspirado e biópsia de medula óssea, eletrólitos, estudo radiológico do esqueleto, entre outros. Alguns pacientes, entre 5% a 32%, desenvolverão manifestações sistêmicas de MM em alguns anos, o que exige acompanhamento prolongado19. O tratamento é cirúrgico com RxT complementar. PEM é reconhecidamente radiossensível, havendo controle regional em mais de 80% dos casos17,19.
Hemangiopericitoma Sinonasal (HPCSN)
O HPCN é um tumor mesenquimal raro da região nasossinusal caracterizado por um padrão de crescimento perivascular proeminente20. Desde sua descrição inicial em 1942, a definição desta patologia como uma entidade clínica específica tem sido questionada21. Apesar da terminologia, atualmente acredita-se que HPCSN é uma neoplasia clínica e patologicamente distinta do hemangiopericitoma de tecidos moles convencional (HPC)20,21. Alguns autores o relacionam com o tumor glômico, considerando-o uma variante deste ou um híbrido entre tumor glômico e HPC20, devido às semelhanças de comportamento biológico, perfil citológico e imunofenótipos.
Entre 15% e 30% de todos os HPC ocorrem na região da cabeça e pescoço e representam menos de 1% das neoplasias vasculares21,22. O HPCSN acomete preferencialmente pacientes na sexta e sétima décadas de vida de ambos os sexos20,21. O quadro clínico mais comum é de obstrução nasal unilateral e/ou epistaxe, com massa de aspecto polipóide na cavidade nasal20,21. Estudo radiológico revela opacificação por massa polipóide, às vezes associada à erosão óssea e esclerose21.
Histologicamente, HPCSN apresenta-se como uma lesão submucosa, não-capsulada, com padrão de crescimento difuso e hialinização perivascular proeminente20,21. Exibe uma aparência mióide sem pleomorfismo celular, com células tumorais organizadas em lâminas ou fascículos20,21. Não apresenta marcadores imunohistoquímicos específicos. Com freqüência há expressão de vimentina, actina e actina de músculo liso20,21. Ao contrário do HPC de tecidos moles, não expressa CD3420,21. No caso apresentado, as células tumorais expressavam apenas vimentina.
Em geral, HPCSNs são indolentes. Tumores de comportamento agressivo, como no caso apresentado, são incomuns, havendo relatos na literatura de lesões com potencial destrutivo local e metástases21. Características histológicas que podem estar associadas a comportamento agressivo incluem maior número de figuras mitóticas, aumento de celularidade com pleomorfismo celular e necrose21. Lesões grandes (maiores ou iguais a 6,5cm) e primeira ressecção incompleta também estão relacionadas a pior prognóstico21.
O tratamento é cirúrgico, variando de polipectomia simples a ressecção ampla com margens livres20,21. Recorrência local ocorre em 17% a 40%21 dos casos, provavelmente devido a tumor residual. O prognóstico é favorável, com taxa de sobrevida livre de doença em 5 anos de 74,2% e em 10 anos de 64,4%21. RxT e QT podem ser usadas para doença metastática e para tumor residual ou primário irressecáveis21.
Neurofibroma
Tumores neurais benignos são responsáveis por cerca de 45% dos tumores de cabeça e pescoço23. O neurofibroma, neoplasia originada das células de Schwann, é o tipo mais comum, acometendo com maior freqüência a pele23. Cerca de 25% a 45% dos neurofibromas ocorrem na região da cabeça e pescoço, sendo que somente 4% acometem fossa nasal e seios paranasais24.
Apresentam-se como lesão isolada ou tumores múltiplos, como ocorre na neurofibromatose tipo 1 (NF1) ou Doença de Von Recklinghausen23. Quase todos os pacientes com NF1 desenvolverão neurofibroma em algum momento de suas vidas25. O potencial de malignização é de 2,6%, sendo que na síndrome varia de 3% a 15%24.
São lesões bem delimitadas, apesar de não-capsuladas. À microscopia, apresentam proliferação de todos os elementos do nervo periférico, dispersos em padrão frouxo e desordenado em estroma mixóide. Predominam células de schwann com núcleos fusiformes23. A arquitetura desordenada ajuda a diferenciá-lo de schwannomas. O tratamento é cirúrgico24,25.
Fibroma Cemento-Ossificante
FCO é um tumor mesodérmico não-odontogênico raro26,27. De acordo com a classificação da World Health Organization, é considerado uma variante do fibroma cementificante, que representa um subgrupo dos cementomas27. Os cementomas, lesões fibro-ósseas que contêm cemento, são subdivididos em quatro subgrupos de acordo com suas características clínicas, histopatológicas e radiológicas: cementoblastoma benigno, fibroma cementificante, displasia periapical cemental e cementoma gigantiforme26,28.
Com etiopatogenia ainda controversa, acredita-se que a origem dessas lesões seja da membrana periodontal, que contém células blásticas capazes de formar osso, tecido fibroso e cemento26. Extração dentária ou trauma têm sido apontados como possíveis fatores causais26.
A grande maioria ocorre na região molar e pré-molar da mandíbula29. Pode acometer também a maxila e, mais raramente, outros seios paranasais. Alguns estudos mostram predileção pelo sexo feminino, sendo mais freqüente na 4a e 5a décadas de vida29. Geralmente, apresenta-se como uma massa assintomática, bem delimitada e de crescimento lento, diagnosticada em estudo radiológico de rotina29. Pode estar associada a dor local, perda de dentes ou alteração de oclusão dentária29. Lesões maxilares grandes podem ocasionar assimetria de face, rinorréia, proptose e diplopia29. Há relatos de tumores agressivos, especialmente quando localizados em maxila e etmóide26.
Radiologicamente, apresenta-se como uma massa bem delimitada, com densidade de partes moles e focos de ossificação, podendo haver destruição óssea30. À microscopia, há tecido celular fibroso com trabéculas ósseas lamelares esferoidais, permeando um estroma fibroso em associação próxima com fibroblastos. Massas de material basófilo acelular são demonstradas como linhas concêntricas, identificadas como cemento30.
O tratamento é cirúrgico, havendo relatos de remissão após ressecção incompleta26,29. RxT é, a princípio, contra-indicada, devido à resistência tumoral e à possibilidade de malignização29,30.
CONCLUSÃO
Os tumores não-epiteliais da cavidade nasal e seios paranasais são patologias raras, com etiopatogenia, comportamento clínico, tratamento e prognóstico extremamente variados. A inespecificidade dos sintomas costuma retardar o diagnóstico. Em vista disso, o profissional deve estar atento à diversidade de patologias que podem acometer essa região, sendo o diagnóstico histopatológico imprescindível para a condução adequada de cada caso.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Cahali BM, Sennes LU, Murakami MS, D'Antonio WE, Ikino CM, Santoro PP et al. Tumores nasossinusais malignos: espectro de apresentação nos últimos 15 anos. Rev Bras Otorrinolaringol 1999;65(2):296-300.
2. Valle A. Epidemiologia do câncer de cabeça e pescoço no Brasil e no mundo. Em: Tratado de otorrinolaringologia. 1ª ed. São Paulo: Roca; 2002. 1: 860.
3. Theilgaard SA, Buchwald C, Ingeholm P, Larsen SK, Eriksen JG, Hansen SH. Esthesioneuroblastoma: a Danish demographic study of 40 patients registered between 1978 and 2000. Acta Otolaryngol 2003;123(3):433-9.
4. Svane-Knudsen, Jorgensen KE, Hansen O, Lindgren, Marker P. Cancer of the nasal cavity and paranasal sinuses: a series of 115 patients. Rhinology 1998;36(1):12-4.
5. Lund VJ, Howard D, Wei W, Spittle M. Olfactory neuroblastoma: past, present and future? Laryngoscope 2003;113(3):502-7.
6. Dulguerov P, Calcaterra T. Esthesioneuroblastoma: the UCLA experience 1970-1990. Laryngoscope 1992;102(8):843-9.
7. Dulguerov P, Allal AS, Calcaterra TC. Esthesioneuroblastoma: a meta-analysis and review. Lancet Oncol 2001;2:683-90.
8. Ferlito A, Rinaldo A, Rhys-Evans P. Contemporary clinical commentary: esthesioneuroblastoma: an update on management of the neck. Laryngoscope 2003;113(11):1935-8.
9. Oda D. Alternative treatment for central giant cell "reparative" granuloma. Advancer in Anatomic Pathology 2003;10(2):110.
10. Plontke SK, Adler C, Gawlowski J et al. Recurrent giant cell reparative granuloma of the skull base and the paranasal sinuses presenting with acute one-sided blindness. Skull Base 2002;12(1):9-16.
11. Carlos R, Sedano HO. Intralesional corticosteroids as an alternative treatment for central giant cell granuloma. Oral Surg Oral Med Oral Pathol 2002;93(2):161-6.
12. Kauzman A, Li S, Bradley G et al. Central giant cell granuloma of the jaws: assessment of cell cycle proteins.J Oral Pathol 2004;33(3):170-6.
13. Kauzman A, Li S, Bradley G et al. Cell cycle alterations in central giant cell granuloma of the jaws: a comparison with giant cell tumor of bone. Oral Surg Oral Med Oral Pathol 2003;96(3):299.
14. Regezi JA. Odontogenic Cysts, Odontogenic Tumors, Fibroosseous, and Giant Cell Lesions of the Jaws. Modern pathol 2002;15(3):331-41.
15. Fernandes AM, Podovani JA, Maníglia JP. Plasmocitoma extramedular de nasofaringe: Relato de um caso e revisão da literatura. Rev Bras Otorrinolaringol 1998;64(3):296-8.
16. Arrarte J, Barra M, Fontoura S, Peduzzi F, Junior H. plasmocitoma extra medular de nariz, relato de un caso. An ORL Mex 2000;45(3):118-20.
17. Sahin S, Dulundu H, Coskun H. Nasal plasmocytoma, proceedings of the XV world congress of ORL, head and neck surgery 1993;2:486-88.
18. Robbins SL, Cotran RR, Kumar VK, Collins T. Robbins pathologic basis of disease. 6a ed. Philadelphia: Saunders 1999;763-4.
19. Courtmans I, Pigeolet Y, Hedayat A, Vilain J. Upper airways locations of plasmocytoma. Acta Oto-Rhino-Laryngol Belgica 2000;54(4):487-90.
20. Li X, Hisaoka M, Morio T, Hashimoto H. Intranasal pericytic tumors (glomus tumor and sinonasal hemangiopericytoma-like tumor): Report of two cases with review of the literature. Patho international 2003;53(5):303-8.
21. Thompson L, Miettinen M, Wenig B. Sinonasal-Type Hemangiopericytoma: A Clinicopathologic and Immunophenotypic Analysis of 104 Cases Showing Perivascular Myoid Differentiation. Am J of Surg Patho 2003;27(6):737-49.
22. Sabini P, Josephson G, Yung R, Dolitsky J. Hemangiopericytoma presenting as a congenital midline mass. Arch Oto Head and Neck Surg 1998;124(2):202-4.
23. Souza L, Oliveira J, Freitas T, Carvalho R. Neurofibroma paciniano: relato de um caso raro de localização intra-oral. Rev Bras Oto 2003;69(6):851-4.
24. Silva V, Agra L, Freitas M, Pinheiro S. Neurofibroma nasal: relato de caso e revisão de literatura. Rev Bras Otorrinolaringol 1999;65(2):172-4.
25. Packer R, Gutmann D, Rubenstein A et al. Plexiform neurofibromas in NF1: Toward biologic-based therapy. Neurology 2002;58(10):1461-70.
26. Jacobs J, Berg H. Destructive cemento-ossifying fibroma of the maxilla. Ear Nose Throat J 1999;69(12):805-8.
27. Bowyer J, Majid M, Ah-Fat F et al. Giant cemento-ossifying fibroma of the maxilla causing proptosis in a young patient. J Pediatr Ophthalmol Strazismus 2001;38(6):359-62.
28. Selmani Z, Anttila J, Mertakorpi J et al. Cemento-ossifying fibroma of the etmoidal sinus in a child presenting with isolated pain in the nasal region. J Craniofacial Surg 2004;15(2):215-7.
29. Sciubba J, Goldstein M, Cohen A, Abramson A. A destructive maxillary cemento-ossifying fibroma following maxillofacial trauma. Laryngoscope 1984;94(6):810-50.
30. Fujimoto Y, Katho M, Miyata M et al. Cystic cemento-ossifying fibroma of the etmoidal cells. J Laryngol Otol 1987;101(9):946-52.
1 Residente de ORL do HGF SESA/SUS.
2 Preceptora da Residência de Otorrinolaringologia do Hospital Geral de Fortaleza, Otorrinolaringologista.
3 Professor Assistente da Disciplina de Patologia Geral da Faculdade de Medicina da Universidade Federal do Ceará, Patologista.
4 Residente de ORL do HGF SESA/SUS.
5 Residente de ORL do HGF SESA/SUS.
Hospital Geral de Fortaleza - SESA/SUS.
Endereço para correspondência: Rua Fausto Cabral 95 apto. 701 Bairro Vicente Pinzon 60175-415 Fortaleza CE.
Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) da RBORL em 31 de março de 2005. cod. 204.
Artigo aceito em 24 de maio de 2005.
Imprimir: ![]()