

Year: 2002 Vol. 68 Ed. 3 - (20º)
Relato de Caso
Pages: 425 to 429
 PDF PT
PDF PT Sinusonasal polyposis in a child with Peutz-Jeghers syndrome
Author(s):
Adriana C. Perez-Bóscollo(1),
Luís C. Carmo(2),
Maria A. Tomaz(2),
Elina T. Oliveira(3),
Ana L. S. Boscolo(4)
Keywords: Peutz-Jeghers syndrome, nasal polyposis, chronic sinusitis, hamartomas, child
Abstract:
Peutz-Jeghers syndrome (PJS) is an autosomal dominant disease, characterized by in association with characteristic mucocutaneous pigmentation. Peutz (1921), in his first publication about the syndrome that has his name, presented two cases having polyps in the nasopharynx. Ever since, a few of those cases were published, as time goes, by association was forgotten. This report describes a rare variant of PJS in a fourteen years-old boy, identified by the presence of bilateral nasal polyposis, chronic sinusitis and hamartomatous intestinal polyposis, in an operated patient previously by intestinal oclusion. At macroscopy, multiple white formations were found, with soft consistency and cystic cavities. Microscopically, the polyps showed inflammatory characteristics associated with atypical escamous metaplasia. In pediatric population, nasal polyps present many matters of specific interest. It is an infrequent condition that requires careful diagnoses work up. The objective of this work is to show a rare disease (PJS) with association in Pediatric Surgery and ORL, calling attention of surgeons, pediatricians and specialists to the importance of investigating the etiology of nasal polyposis in the pediatric patients.
![]()
INTRODUCTION
Peutz-Jeghers syndrome (PJS) was described for the first time by a London surgeon in 1896, Jonathan Hutchinson6. However, the association of heredity in the syndrome was first recognized by Peutz (1921) in a German family whose clinical characteristics included gastrointestinal polyposis, mucus-cutaneous polyposis, mucus-cutaneous pigmentation, nasal polyposis and rectal extrusion of polyps11. In 1949, the physicians Jeghers, Mukusiak and Katz noticed the presence of dominant autosomal heritage and the syndrome became known by the scientific community8. PJS is identified based on the triad of skin and membranous mucosa pigmentation, gastrointestinal tract hamartomatous polyposis and family history5, 6. Nasal polyposis was referred by Peutz in his initial observations and since then, few cases have been reported in the specialized literature1, 3, 7, 9, 15.
Publications made in past years described higher morbidity-mortality related to PJS owing to complications of the disease in the gastrointestinal tract2, 3, 4, 14, or higher incidence of cancer in different systems3, 15. The advance in genetic research studies allowed the identification of genes involved in chromosome mutations responsible for malignant transformation of polyps and melanosis 10, 12, 13.
We have recently seen a case of bilateral nasal polyposis of nasopharynx and maxillary sinus in a 14-year old boy, with clinical diagnosis of chronic sinusitis and history of surgical treatment for intestinal obstruction by multiple polyps. The purpose of the present study was to show a rare association between Otorhinolaryngology and PJS that involved surgeons, pediatricians and specialists and to highlight the importance of the diagnosis of nasosinusal polyposis.
CASE REPORT
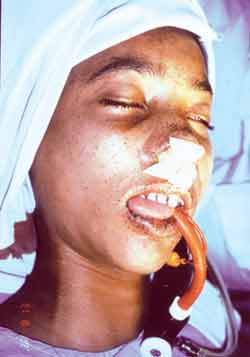
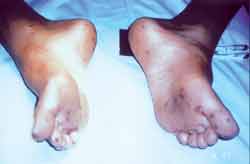
Fourteen year-old Black male subject submitted to jejune-ileal resection six years before caused by intussusception and necrosis of intestinal loop. In the pathology of the surgical specimen, we found multiple polyps that were microscopically classified as hamartoma. The patient also presented significant lip, peri-oral and digital pulp pigmentation (Figures 1 and 2, respectively). Since there were tegument abnormalities in his younger sister and father, we defined the diagnosis of Peutz-Jeghers syndrome.
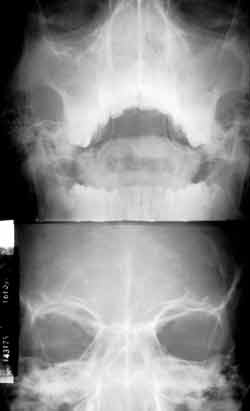
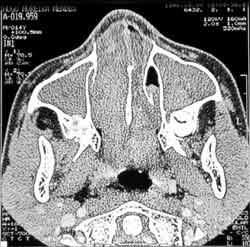
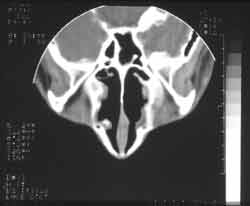
Two years after, the patient presented nasal obstruction, hyaline coriza and night snoring. He visited the pediatrician who diagnosed allergic rhinitis and started clinical treatment with no improvement of symptoms. The patient experienced a number of fever episodes, purulent rhinorrhea, cascomia and continuous respiratory difficulties. Followed up in the discipline of Pediatrics, he was treated of chronic sinusitis for one year. Without response, he was then referred to the discipline of ORL to perform further investigations. At ectoscopy, we observed frontal edema and enlargement of the nasal dorsum and tip. Anterior rhinoscopy showed a large gray mass filling the nasal cavity, little depressive, bilateral and with fetid purulent secretion. Routine laboratory tests did not show abnormalities. Paranasal sinuses x-rays at different incidences showed velamentum of both maxillary sinuses (Figure 3). CT scan did not identify the turbines and showed bilateral nasal polyposis with paranasal sinus velamentum (Figure 4).
Based on these results, we scheduled surgical resection of the polyps by Caldwell-Luc approach. During the surgical act, we observed multiple polyps in the nasopharynx, nasal fossa and maxillary sinuses, gray in color, increased consistency, significant bilateral maxillary mucocele and destruction of lateral nasal walls. We resected the material of the nasopharynx and maxillary sinus and sent it to hystopathology analysis. After the surgery, we prescribed local cleaning with sterile solution at 0.9% and topical rifocine.
Histology
Macroscopy
Nasal fossae and paranasal sinuses: dark white multiple polyps, weighing 120g and measuring all together 8.0x7.5x3.0cm, hardened consistency.
Microscopy
Polyps recovered by respiratory epithelium, showing areas of mature and immature squamous metaplasia. Submucous layer showed marked edema and inflammatory infiltrate formed predominantly by plasmocytes and lymphocytes and by occasional eosinophils. We also noticed cystic dilations filled with mucus and neutrophilic inflammatory infiltrate foci amidst the epithelium.
The patient has been clinically followed up for 10 months and has not presented recurrence of polyps (Figure 5). At present, he presents no complaints and has normal upper airway physiology.
Figure 1.
Figure 2.
Figure 3.
Figure 4.
Figure 5.
DISCUSSION
This is the report of a rare disease (PJS) associated to nasal polyposis in a 14-year old child. The diagnosis of the patient was based on pathological and family history and histopathology of the collected material during nasosinusal surgery, according to the reports referred by the literature5, 8, 11.
Pigmentation affects especially the lips and oral mucosa (94%)6, 8, hands (74%), feet (62%) or any other body part (21%)14. Polyposis can affect any portion of the gastrointestinal tract, but it is more common in the jejune5, which was also observed in our patient. Patients do not become symptomatic up to the 2nd decade of life4, 11, when polyps tend to infarct, ulcerate, bleed or cause intussusception and intestinal obstruction3, 14. At the age of 8, our patient was submitted to jejune-ileal resection because of intussusception and necrosis of intestinal loop. In the study of the surgical piece, they found polyps microscopically classified as hamartomas. The polyps are rarely found in the mouth, esophagus, ureter, gallbladder, renal pelvis, bronchia, nose, maxillary sinus and lungs5. Our patient presented bilateral nasosinusal polyposis with significant clinical repercussion. The confirmation of family history of PJS contributed to the diagnosis, but it is really hard to collect reliable family data. In our case, the investigation of close relatives led to suggestive data: the father and the younger sister had typical pigmentation and complaints that justified the investigation. The susceptibility gene of PJS codified by STK11 (expressed in serin/theorinine kinase, also called LKB1), was identified in families who had the syndrome. Results confirmed the mapping of common locus 19p13.3, but also suggested the existence, in a minority of families, of the potential locus 19q13.410.
The survival of the families is frequently reduced by surgical emergencies such as complications of polyps that generate repetitive surgeries, which may lead to short bowel syndrome2. In addition, PJS is associated to lung cancer, ovarian sexual tumor, malignant adenoma (a malignant form of neck cancer) and female testicle tumors of Sertolli cells in pre-puberty boys3, 15. The identification of mutations of the germinative line of the family could be the initial step for managing the syndrome, because it contributes to the development of sporadic and family forms of cancer12, 13, gastrointestinal tract malignant diseases and extraintestinal tissues15.
Although Peutz had included nasal polyposis in his definition of the syndrome, it has been ruled out because it was thought that nasal polyposis occurred so commonly in the general population that it could not be part of a specific syndrome15. Westerman et al. presented six cases of nasal polyposis found in descendents of a family investigated by Peutz15. One of them developed squamous cell carcinoma of the nasal cavity. It was not clear whether the polyposis of those patients classified as inflammatory polyps could be considered hamartomas of PJS. The association of nasal polyposis and PJS has also been reported by other authors1, 3, 7, 9. Cerqua et al. have advocated the inclusion of nasal polyposis in the clinical picture of the syndrome1. The development of nasal cavity cancer in a patient with PJS and nasal polyposis had not been reported before Westerman publication, even though De Facq had reported adenomatous transformation of two nasal polyps in one patient with PJS 3.
CLOSING REMARKS
Based on what was reported, we would like to remind otorhinolaryngologists, pediatricians and surgeons that PJS is a disease with significant morbidity and mortality and we suggest that nasal polyposis becomes part of its clinical picture. Therefore, this syndrome should be investigated in pediatric patients that have some of the typical characteristics and upper airway disorders that do not respond to the appropriate therapy. Thus, we would be able to perform early diagnosis and contribute to improve the survival of affected families. We insist that new reports should be made to provide further understanding about this rare disease and its ENT associations.
REFERENCES
1. Cerqua N, D'ottavi LR, Perotti V. Rare manifestazioni dell poliposi nasale nella sindrome di Peutz-Jeghers. Acta Otorhinol Ital 1993;13:333-338.
2. Choi HS, Park YJ, Park JG. Peutz-Jeghers syndrome: a new understanding. J Korean Med Sci 1999;14:2-7.
3. De Facq L, De Sutter J, De Man M et al. A case of Peutz-Jeghers syndrome with nasal polyposis, extreme iron deficiency anemia, and hamartoma-adenoma transformation: management by combined surgical and endoscopic approach. Am J Gastroenterol 1995;90:1130-1132.
4. Foley TR, Garrity TJ, Abt AB. Peutz-Jeghers syndrome: A clinicopathologic survey of the "Harrisburg Family" with a 49-year follow up. Gastroenterol 1988;95:1535-1540.
5. Hemminki A. The molecular basis and clinical aspects of Peutz-Jeghers syndrome. Cell Mol Life Sci 1999;55:735-750.
6. Hutchinson J. Pigmentation of lips and mouth. Arch Surg 1896;7: 290.
7. Jancu J. Peutz-Jeghers syndrome involvement of the gastrointestinal and upper respiratory tracts. Am J Gastroenterol 1971;56: 545-549.
8. Jeghers H, Mckusick VA, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits. New Engl JL 1949;241:992-1005.
9. Manegold BC, Bussmann JF Fürstenberg HS. Klinischer beitrag zum Peutz-Jeghers-syndrom mit befall des magendarmtraktes, der oberen luftwege sowvie beider mammae. Med Welt 1969;25:1435-1439.
10. Mehenny H, Blouin JL, Radhakrishna U et al. Peutz-jeghers syndrome: confirmation of linkage to chromosome 19p13.3 and identification of a potential second locus, on 19q13.4. Am J Hum Genet 1997;61:1327-34.
11. Peutz JL. A very remarkable case of familial polyposis of the mucous membrane of the intestinal tract and nasopharynx accompanied by peculiar pigmentation of the skin and mucous membrane. Ned Tijdschr Geneeskd 1921;10:134-146.
12. Su GH, Hruban RH, Bansal RK et al. Germiline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am J Pathol 1999;154:1835-40.
13. Trojan J, Brieger A, Raedle J et al. Peutz Jeghers syndrome: molecular analysis of a three-generation kindred with a novel defect in the serine theorinine kinase gene STK11. Am J Gastroenterol 1999;94:257-61.
14. Utsunomiya J, Gocho H, Miyanaga T et al. Peutz-Jeghers syndrome: Its natural course and management. John Hopkins Med J 1975;136:71-82.
15. Westerman AM, Entius MM, De Baar E et al. Peutz-Jeghers syndrome: 78-year follow-up of the original family. Lancet 1999;353:1211-5.
[1] Joint Professor, Ph.D., Discipline of Pediatric Surgery, Medical School, Triângulo Mineiro (FMTM).
[2] Undergraduates, Medical School, FMTM.
[3] Pediatric Surgeon.
[4] Otorhinolaryngologist.
Address correspondence to: Dra. Adriana Cartafina Perez Bóscollo - Disciplina de Cirurgia Pediátrica - Departamento de Cirurgia - Hospital Escola - Faculdade de Medicina do Triângulo Mineiro
Rua Getúlio Guaritá nº 130 - Bairro Abadia, Uberaba, MG, Brasil 38025-440, Tel: (55 34)318.5228 - Fax: (55 34)312.2390 - E-mail: boscollo@mednet.com.br
Study presented at XXI Congresso Brasileiro de Cirurgia Pediátrica, October 2000.
Porto Alegre - RS - Brazil.
Print: ![]()