

Year: 2004 Vol. 70 Ed. 2 - (6º)
Artigo Original
Pages: 182 to 186
 PDF PT
PDF PT Genetic investigation of non-syndromic hereditary deafness
Author(s):
Leopoldo N. Pfeilsticker 1,
Guita Stole 2,
Edi Lucia Sartorato 3,
Denise Delfino 4,
Andréa Trevas Maciel Guerra 5
Keywords: hereditary hearing loss, genetic, conexin26
Abstract:
The precise diagnosis of hearing loss can be clarified by genetic investigation. Non-syndromic hearing loss is responsible for 70% of all genetic causes of hearing loss. More than 100 genes are potentially involved in non-syndromic hearing loss. A specific mutation (35delG) on the GJB2 gene that codifies Conexin 26 protein is the most common finding in non-syndromic hereditary hearing loss. Aim: In this study the presence of mutations 35delG, A1555G/12SeRNA and A7445G/tRNASer (UCN) where investigated for patients with hearing loss of unknown cause. Study design: Clinical study with transversal cohort. Material and method: 75 outpatients from the Department of Otolaryngology and Head and Neck Surgery of the University of Campinas-UNICAMP were evaluated from July to December of 2000. A total of six mutations were found, four 35delG/GJB2, one A7445G/tRNASer (UCN) and W172X/GJB2, a mutation not yet described in previous literature. Conclusion: The investigation of mutations associated with hearing loss can be carried out easily, elucidates the etiology and allows genetic counseling of the family.
![]()
INTRODUCTION
The study of genetic deafness lists many diagnostic possibilities. Many patients with sensorineural loss of unknown cause can present chromosome alterations that have determined the pathology.
The conduction of simple and affordable tests to investigate the most common mutations associated with hearing loss has allowed the etiological definition of some cases.
It is estimated that there are over 100 genes that are potentially involved in nonsyndromic hearing loss. After the location of locus DFNA1 in chromosome 5q31 in 1992, about 70 different loci have been mapped. Since the report of the first mutation in a nuclear gene associated with nonsyndromic autosomal deafness in 1997, other 15 genes have been cloned 1, 2.
It is estimated that 70% of all genetic causes of deafness are nonsyndromic and among them, 80% are recessive autosomal hereditary affections. Gene GJB2, which codifies protein connexin 26 (Cx26) is involved in both dominant and recessive forms of nonsyndromic deafness. Mutations of this gene are responsible for 80% of the cases with recessive hereditary pattern and one specific mutation (35delG) is the main one involved in genetic etiology deafness cases: it is loss of guanine base in the DNA sequence, at position 35. This mutation amounts to 75 to 80% of al mutations found in this gene. 3, 4
Mutation A1555G in mitochondrial gene 12SrRNA was the first one to be associated with nonsyndromic deafness and defined the standard of maternal hereditary transmission 5. The mutation has been associated with deafness related to ototoxicity of aminoglycoside and in the United States it is presented in 15% of all patients with antibiotic-induced deafness. Another mitochondrial mutation that defines deafness is A7445G in gene tRNASer (UCN), which is associated with palmoplantar keratodermia 6,7.
OBJECTIVES
To investigate the presence of mutations 35delG/GJB2, A1555G/12SrRNA and A7445G/tRNASer (UCN) in patients whose etiological diagnosis of hearing loss had not been determined or who had positive indication in their genetic history. To assess the influence of this information to define the etiology of hearing loss.
MATERIAL AND METHODS
We included in the study patients seen by the Ambulatory of Otoneurology and Communication Disorders, Discipline of Otorhinolaryngology and Head and Neck Surgery, State University of Campinas - UNICAMP, from July to December 2000, which met at least one of the following criteria:
1. Hearing loss without defined etiological diagnosis;
2. Hearing loss not proportional to the identified cause;
3. Early hearing loss;
4. Positive family history or presence of consanguinity.
Patients that met the criteria were submitted to genetic assessment, regardless of the conduction of other indicated tests (metabolic profile, autoimmune profile, serologic tests, high resolution imaging tests).
Blood samples were collected after patients had understood the objectives of the test and signed the informed consent term. They were sent to the Laboratory of Molecular Biology and Genetic Engineering of State University of Campinas (CBMEG) so that investigation of mutations of Cx26 35delG and mitochondrial mutations A1555G and A7455G in genes 12SrRNA and tRNASer (UCN), respectively, could be conducted. Next, we used sequencing to search for other mutations of GJB2. Once any alteration was identified, clinical follow-up was also conducted by the Department of Genetics, Medical School, UNICAMP.
All DNA samples were screened to detect presence of mutations of 35delG using allele-specific polymerase chain reaction (AS-PCR), but with the modification patented by CBMEG, UNICAMP. The method can easily discriminate the normal allele from the mutant one and by means of two reactions it is possible to differentiate normal homozygous, 35delG homozygous and heterozygous with mutation 8.
In order to investigate mitochondrial mutations, we used PCR technique to amplify two regions of mitochondrial DNA of the patients, one containing nucleotide 1555 and the other containing nucleotide 7445, according to the mitochondrial sequencing published by Cambridge 9.
RESULTS
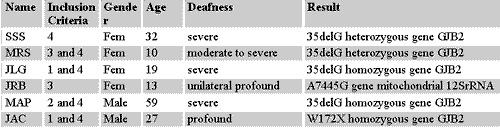
Out of 75 patients that met the inclusion criteria, there were six mutations, being that 4 had mutations of 35delG (Chart 1). There were two homozygous and two heterozygous with the mutation. As to the other two mutations, one affected GJB2 (homozygous W172X/W172X), which is a mutation that had never been described. The other one (A7445G) was identified in mitochondrial gene 12SrRNASer (UCN). In 19 patients, total sequencing of GJB2 was completed and 14 of them were normal. In 56 patients we did not find the presence of 35delG, but other mutations, related or not with Cx26, could still be found. Seventy-three patients did not have the studied mitochondrial mutations.
Case 1
MRS, female, 10 years, bilateral progressive moderate/severe sensorineural loss for 3 years, followed up during 8 months with diagnosis of cochlear otosclerosis. Initial assessment was based on family history of maternal hearing loss, also considered to be otosclerosis. Result of genetic assessment: heterozygous for 35delG.
Case 2
MAP, male, 59 years, bilateral severe sensorineural loss since childhood, with history of otorrhea since then. Three siblings with hearing loss. Result of genetic assessment: homozygous for 35delG.
Case 3
SSS, female, 32 years, bilateral severe sensorineural loss with consanguineous parents, brother and daughter with hearing loss. Followed up for almost 4 years with diagnosis of cochlear otosclerosis. Result of genetic assessment: heterozygous for 35delG.
Case 4
JLG, female, 19 years, bilateral severe sensorineural loss since childhood with family history of 3 siblings with complete deafness. Result of genetic assessment: homozygous for 35delG.
Case 5
JAC, male, 27 years, bilateral severe to profound sensorineural loss since childhood. Nine siblings, 3 with deafness, being that 2 of them had been deaf since childhood. Result of genetic assessment: homozygous for W172X.
Case 6
JRB, female, 15 years, profound hearing loss on the left, mild to moderate loss on the right. Maternal rubella and positive serology at birth, according to the mother. History of repetitive otitis and use of bilateral ventilation tubes. Result of genetic assessment: A7445G mitochondrial gene 12SrRNA.
DISCUSSION
Connexins are proteins that form the intercellular junctions, the key site for communication between cells for exchange of electrolytes, secondary messengers and metabolites. Cx26 has oligomeric bounds to other five identical (homotypical) or non-identical (heterotypical) units forming hexomers called connexons. They are immersed in plasmatic membrane and form disulfide bridges with connexons of neighboring cells, facilitating the direct transfer of small molecules from cytoplasm to cytoplasm 10. In the inner ear, Cx26 influences functioning of stria vascularis, basilar membrane, limbus and spiral prominence of human cochlea. Loss of connexin function in this complex contributes to changing permeability of supporting cells and fibroblasts of Corti's organ, allowing large concentrations of potassium present in the cochlear duct to diffuse to the organ, modifying the transport and permeability of the ion in the hair cells' synapses, leading to hearing loss 11.
In a microscopic assessment of the temporal bone of a patient with deafness that presented heterozygous mutation of 35delG, Jun et al. evidenced absence of neural degeneration, preserved spiral ganglion cells, almost total degeneration of hair cells of Corti's organ, disconnected tectorial membrane, vascular stria agenesia, and large cysts of scala media in the stria vascularis region 12.
The correlation between specific mutation of 35delG and severity of progression of hearing loss has not been defined. Cohn et al. did not find any consistent auditory phenotype of affections to locus DFNB13. Rabionet et al. suggested that these variations would indicate participation of environmental factors 13. Park et al. proposed that differentiated manifestations of mutations of Cx26 would be attributed to racial factor 14. Thus, even though the identification of these anomalies provide important information for those that have deafness, it is important to be very careful whenever stating things about genetic variation if there is no demonstrable disease. However, if there is 25% risk of recurrence of genotype, counseling is essential.
Even though genotype does not predict hearing, there is significant incidence of homozygous 25delG in pre-lingual deafness: 26 to 30% will have severe deafness and the other 30 to 57% will have profound deafness. Severe to profound hearing loss will affect approximately 1 to 1,000 neonates. In 50% of these children, the loss is presumably genetic, received through recessive autosomal hereditary. For this reason, it is suggested that genetic test, owing to its low cost, should be included in the battery of tests for the investigation of sensorineural hearing losses 4, 11, 12.
In our study, four patients presented positive genetic tests for 35delG, being that two were heterozygous, two were homozygous and they all presented severe hearing loss.
The presence of other morbid conditions can change the clinical status of patients modifying the manifestations of the genetic disease. Out of six mutant patients, one (SSS) presented compensated diabetes mellitus type 2, and the family history of another one (JRB) masked the genetic origin of the hearing loss.
Some authors believe that other mutations involving Cx26 can be related to pathophysiology of ear degenerative diseases and would explain progressive hearing loss such as presbyacusis 15.
Many genes associated with hereditary deafness, contrarily to what was believed, do not express in hair cells but rather in supporting cells distributed throughout the cochlear duct. The discovery of mutations and their effects on hearing showed that the role of these cells on cochlear function is indispensable 1.
Even though there is still some controversy, the involvement of GJB2 in autosomal dominant deafness has also been proposed in a small percentage of these cases (there were 5 mutations described) 16-18.
Few papers relating mutations associated with hearing loss and vestibular function have been published. The patients described in this study did not present clinical vestibular abnormalities. Kunst et al. described some instrument vestibular findings of nonspecific characteristics and without identifiable pattern 2, 12, 13.
The incidence of mutations of gene GJB2 in the world literature is superior to the data obtained in our study, which is probably a result of the strict selection criteria and the fact that we did not conduct complete sequencing of all genes in all patients of the study.
Mutation homozygous W172X as the one observed in patient JAC had never been reported in the literature. It was confirmed by analysis of restriction and causes the exchange of triptophane amino acid for terminal codon, clearly interfering in protein functioning. This mutation has been described only in this Brazilian patient with probable history of family consanguinity.
Mitochondriae are intracell organelles responsible for the production of most of the energy produced in the cells. They have small genomic content with maternal pattern of heritage, and if mutant they can originate a wide range of diseases 19. Mutations of mitochondrial DNA are associated with nonsyndromic and syndromic deafness. Environmental factors such as aminoglycoside antibiotics, as well as probable non-identified nuclear genes, interact with these mutations and determine the expression of auditory phenotypes 19, 20.
We described 5 mutations that determined nonsyndromic hearing loss. In two of these mutations (7445G, 7472C), in addition to hearing loss, other symptoms were present. It is not known why these five mutations preferably affect the inner ear, despite the essential role that mitochondria play in almost all cells of the human body 19.
Phenotypic manifestation of the diseases resultant from DNA mitochondrial alterations is normally associated with late start, selectivity of the affected organ, and progressive evolution of crisis 21.
CONCLUSION
Screening for genetic mutations associated with hearing loss is easy and affordable.
Mutations 35delG of Cx26 are potentially related with some cases of undefined hearing loss. The study of these mutations should be included in the battery of tests for etiological investigation of undetermined deafness, since it helps the understanding of the case and allows genetic counseling in case of positive results.
In patients in which there is great suspicion of hereditary disease, we should progress with the study of other genes.
Chart 1. Mutations found in the genetic investigations.
REFERENCES
1. Steel KP, Bussoli TJ. Deafness genes, expressions of surprise. TIG 1999; 15(6):207-11.
2. Kunst H, Marres H, Huygen P, Van Duijnoven G, Krebsova A, Van Der Velde S, Reis A, Cremers F, Cremers C. Non-syndromic autosomal dominant progressive non-specific mid-frequency sensorineural hearing impairment with childhood to late adolescence onset (DFNA21). Clin Otolaryngol 2000;25:45-54.
3. Cohn ES, Kelley PM, Fowler TW et al. Clinical studies of families with hearing loss attributable to mutations in the connexin gene (GJB2/DFNB1). Pediatrics 1999; 103(3): 546-50.
4. Cohn ES, Kelley PM. Clinical phenotype and mutations in connexin 26 (DFNB1/GJB2), the most common cause of childhood hearing loss. Am J Med Genet 1999; 89:130-6.
5. Jaber L, Shohat M, Bu X, Fischel-Ghodsian N, Yang HY, Wang SJ, Rotter JI. Sensorial deafness inherited as a tissue specific mitochondrial disorder. J Med Genet. 1992; 29:86-90.
6. Fischel-Ghodsian N, Prezant TR, Fournier P, Stewart IA, Maw M. Mitochondrial tRNA mutation associated with non-syndromic deafness. Am J Otolaryngol 1995; 16: 403-8.
7. Sevior KB, Hatamochi A, Stewart IA et al. Mitochondrial A7445G mutation in two pedigrees with palmoplantar keratoderma and deafness. Am J Med Genet 1998; 75:179-85.
8. Sartorato CA, Gottardi E, Oliveira CA et al. Determination of carrier frequency of the 35delG mutation in Brazilian neonates. Clinical Genetics 2000; 58(4): 339.
9. Anderson S, Bankier AT, Barrel BG et al. Sequence and organization of the mitochondrial genome. Nature 1981; 290:457-65.
10. Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem 1996; 238(1):1-27.
11. Lefebvre PP, Van De Water TR. Connexins, hearing and deafness: clinical aspects of mutations in the connexin 26 gene. Brain Research Reviews. 2000; 32:159-62.
12. Jun AI, Mcguirt WT, Hinojosa R, Green GE, Fischel-Ghodsian N, Smith RJH. Temporal bone histopathology in Connexin 26 related hearing loss. Laryngoscope 2000; 110:269-75.
13. Rabionet R, Gasparini P, Estivill X. Molecular genetics of hearing impairment due to mutations in gap junction genes encoding beta connexins. Human Mutations 2000; 16:190-202.
14. Park HJ, Hanh SH, Chun YM, Park K, Kim H. Connexin 26 mutations associated with nonsyndromic hearing loss. Laryngoscope 2000; 110:1535-8.
15. Morell RJ, Kim HJ, Hood LJ et al. Mutations in the connexin 26 gene (GJB2) among Ashkenazi jews with nonsyndromic recessive deafness. N Engl J Med 1998; 339:1500-5.
16. Denoyelle F, Lina-Granade G, Plauchu H et al. Connexin 26 gene linked to a dominant deafness (letter). Nature 1998; 393:319-20.
17. Scott DA, Kraft ML, Stone EM, Sheffield VC, Smith RJ. Connexin mutations and hearing loss (letter). Nature 1998; 391:32.
18. The Connexin-deafness homepage. http://www.iro.es/cx26.htm
19. Van Camp G, Smith RJ. Maternally inherited hearing impairment. Clin Genet 2000 Jun; 57(6):409-14.
20. Jacobs HT. Mitochondrial deafness. Ann Med 1997 Dec; 29(6):483-91.
21. Guan MX, Fischel-Ghodsian N, Attardi G. Biochemical evidence for nuclear gene involvement in phenotype of non-syndromic deafness associated with mitochondrial 12S rRNA mutation. Hum Mol Genet 1996 Jul; 5(7):963-71.
1 Assistant Physician, Division of Otoneurology and Skull Base, Discipline of Otorhinolaryngology and Head and Neck Surgery, UNICAMP.
2 Assistant Physician, Division of Otoneurology, Discipline of Otorhinolaryngology and Head and Neck Surgery, UNICAMP.
3 Ph.D. in Human Genetics - UNICAMP-CBMEG.
4 Former Resident Physician, Discipline of Otorhinolaryngology and Head and Neck Surgery, UNICAMP.
5 Ph.D. in Human Genetics - UNICAMP- Medical School, Cidade Universitária ZeferinoVaz.
Study presented at II Congresso Triológico de Otorrinolaringologia, held in Goiania in 2001, awarded with special citation.
Discipline of Otorhinolaryngology and Head and Neck Surgery - UNICAMP
Address correspondence to: Cidade Universitária Zeferino Vaz CP 6111, Barão Geraldo, Campinas, SP 13083-970
Tel (55 19) 3788-7523 - E-mail: lnp@uol.com.br
Print: ![]()