

Year: 2001 Vol. 67 Ed. 1 - (17º)
Relato de Casos
Pages: 107 to 112
Genetic Syndromes Associated with Hypoacusis. Three Cases Report and Literature Review.
Author(s):
Daniel C. Pinheiro*,
José F. Colafêmina**,
Aloysio A. T. C. Netto*,
João M. Pina Neto***.
Keywords: Goldenhar syndrome, hereditary diseases, genetics, medical, otorhinolaryngologic diseases, hearing loss sensorineural
Abstract:
The congenital hypoacusis may follow syndromes defined by abnomalities in several organs and systems. In some cases, the hearing loss may be the first symptom of a non discovered pathology, where the doctor must be aware of the possibility of others problems associated to this symptom. The number of genetic syndromes associated with hearing loss is so big. We describe the most usual with their clinical characteristics, and we report three cases in the Otology Service of Clinics Hospital of Medicine School of Ribeirão Preto, in São Paulo University (Brazil).
![]()
INTRODUCTION
Congenital hearing loss may accompany syndromes characterized by deficits in different organs and systems. In some cases, hearing loss may be the first symptom of an unknown pathology and the Otorhinolaryngologist should be attentive to the fact that other signs could be associated with the hearing complaint'. It is estimated that 50% of all sensorineural hearing losses in children are caused by genetic factors, and most of them are transmitted by means of recessive genes 2,3,7. This figure is alarming because there are no effective treatment approaches for most of these cases. Thus, prevention is the only alternative to reduce the high incidence7.
Genetic hearing losses may be congenital or late, uni or bilateral, monosymptomatic or syndromic. Monosymptomatic syndromes are the most common, accounting to 70% of congenital sensorineural hearing losses', but they are more difficult to be diagnosed 2,7.
Association of genetic syndromes and hearing loss is very common7. The present study aimed at reporting 3 cases treated at the Ambulatory of Otology at Hospital das Clínicas, Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, and providing a brief review of the literature.
Goldenhar's syndrome
Many terms have been used to name this entity: hemifacial microsomy, ocular-auricular-vertebral dysplasia, syndrome of Goldenhar-Gorlin, syndrome of the first and second brachial arches, and lateral facial dysplasia. It is accepted that the term ocular-auricular-vertebral spectrum is the most correct one 4. It is caused by multifactorial heritage, with positive family history in 6% of the cases.
It is a complete malformation, usually unilateral, involving the structures that derive from the first and second brachial arches. Facial asymmetry is present in 65% of the cases, which become more evident with age. Maxillary, temporal and malar bones on the affected side are usually smaller and thinner. In 10% to 30% of the patients involvement is bilateral, usually displaying one side more affected than the other, more frequently on the right side. There may also be, in some cases, agenesis of salivary glands 1, 4. Ocular abnormalities are common. Epibulbar dermoid is present in 35% of the cases. Blepharoptosis and narrowing of palpebral fissure occur in 10% of the patients. Anophthalmia and microphthalmia have been reported in subjects with severe affection and they may related to mental disability. Facial nerve paralysis is present in 10% of the individuals probably caused by bone impairment at the level of Fallopian canal.¹
It is estimated that the mental deficit is present in 5% to 15Q/o of the affected subjects and central nervous system malformations may also be identified, such as hydrocephalus, dermoid cyst, teratoma, malformation of Alnold-Chiari, arachnoid cyst and hypoplasia of corpus callosum 1,4.
It is also common to find pre-auricular protuberances or fistulae as the first manifestations. Skeletal abnormalities most commonly found are in the cervical vertebrae, such as occipitalization of atlas, synostosis and cuneiform vertebrae. There are also structural alterations of temporal bone that predispose meningitis. Recent findings report the association with cardiac impairment, such as Fallot tetralogy and ventricular septum defect; pulmonary deficit, such as incomplete lobulation, pulmonary hypoplasia or agenesis; renal deficits, such as renal agenesis, double ureter, vascular abnormalities, hydronephrosis and hydroureter; gastrointestinal system compromise, such as anal imperforation with or without rectovaginal fistula 1,4,5.
Both conductive and sensorineural hearing losses have been reported in more than 50% of the cases. The etiology of the loss may vary and present middle and external ear abnormalities: hypoplasia of ossicle chain, aberrant facial nerve, abnormalities of Eustachian tube and skull base4.
Prognosis depends on phenotype of child and presence or not of mental disability4.
Stickler's syndrome
It is a autonomic dominant transmission entity with variable expression, estimated to occur in 1 in each 20,000 people, also known as Marshall-Stickler's syndrome and Wagner-Stickler's syndrome or hereditary artro-ophthalmopathy. The most common characteristics are flat face, cleft palate, severe myopia with retina detachment and cataract, hearing loss and arthropathy with spondiloepiphysial dysplasia. Craniofacial manifestations may vary from normal (15% to 25%) to alterations such as short maxilla, prominent eyes, epicanthal folds, depression of nasal dorsum, and small chin. Cleft palate and abnormalities of palate mobility are reported in 20% of the cases', 4, 9,'0. Myopia that may reach 18 diopters is found in 75% to 80% of the patients. The onset is normally early, before 5 years of age. Corium retina and vitreum degeneration takes place. before 24 years of age in 70% of the cases and it is frequently bilateral. If alterations are not treated early, they may progress to blindness. Total detachment of retina is present in more than half of the patients, which may happen spontaneously or during cataract surgery. Other commonly found alterations are astigmatism (60%), cataract (45%), strabismus (30%) and glaucoma (10%)4.
Joints are normally expanded and frequently present hyper-extensibility, sometimes with pain and heat. Progressive degeneration of joints is described in 30% of the affected patients, sometimes in patients younger than 30 years of age. Mitral valve prolapse is found in about 50% of the cases'. Sensorineural hearing loss is more marked in high frequencies and with a progressive nature, present in 80% of the patients 1, 4.
Marshall's syndrome
It is a autonomic dominant transmission disorder of variable expression. Myopia, cataract, large base nose, sensorineural hearing loss and hypertelorism are the main characteristics of the syndrome. Failure of sight takes, place, in the second decade of life, but there are cases in which loss of sight happened within the first 6 months, and myopia may be higher than 10 diopters. There are also reports of cases of retina detachment. All patients present severe depression of nasal dorsum and anteverted nostrils 1, 4.
Sensorineural hearing loss is normally more marked in high frequencies and starts during childhood. It may be progressive and of late onset in many cases, without affecting the vestibule 1,4.
Radiological study may show hypoplasia or even absence of nasal bones, maxillary hypoplasia and absence of frontal sinus. There are also intracranial calcifications, vertebral malformations, irregular and small pelvis, delay in closure of pubic and ischial bones, medialization of ulna and radio, and in some cases, epiphysial irregularities of extremities 4. CT scan of temporal bones usually reveals normal inner ears and in some individuals narrowing of internal ear canal. Lack of development of ethmoid bone may cause the onset of a smaller anterior cranial fossa¹.
Genetic counseling is useful to prevent new cases. Plastic surgery may bring some benefits for the correction of the nose. Cataract may be corrected with surgery and hearing loss should be minimized with hearing aids and special education1.
Case 1 - Goldenhar's syndrome
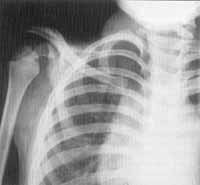
G. A. S., Caucasian female 11-year-old patient, referred to the Service of Olorhinolaryngology at Hospital das Clínicas for audiological assessment, followed by the Sector of Human Genetics because of facial asymmetry since birth and definite diagnosis of Goldenhar's syndrome. The mother reported that the child was healthy except for frequent otalgias and "chest stridor"; she did not refer hearing loss or delay in mental or language development. Parents did not have consanguinity and there was no history of abortion or congenital diseases in the family. The other child of the couple had perfect physical and mental health. The physical exam revealed asymmetric face, underdeveloped left hemiface, brow area on the left lower than on the right, and microphthalmia on the left. Large base nose, with enlarged left nostril. Poorly developed buccal rhyme, deviated to the left. Brachycephaly, bowed frontal bone, and low implantation of hairline. Short neck with normal mobility. Irregular helix, with sulcus on the horizontal region. Peripheral facial paralysis grade IV of House-Brackman 5 on the left. Normal thorax, inverted nipples, normal genitals, perineum and spine. Syndactylia of the second and third toes. The otoscopic exam revealed opacified and retracted tympanic membranes bilaterally, with normal auditory canal. Oropharyngoscopy revealed palatine tonsils grade II, no hyperemia, secretion or other alteration. Pure tone audiometry showed mild conductive hearing loss on the right at 6 and 8KHz and mixed moderate loss on the left, and type B tympanometric curve bilaterally. Ophthalmologic exam showed coloboma of the optical nerve. Head CT scan showed cranial asymmetry to the left, without other findings. Temporal bone CT scan excluded the suspicion of internal or middle car malformation, showing only velomentum of tympanic cavity, suggesting chronic otitis media without bone erosion. Nasophatynx x-ray showed adenoid hypertrophy narrowing the airways, and spinal x-ray showed spina bifida at L4 and L5, maintaining disk space. We started clinical management of serous otitis media with predinisolone 0.5mg/kg for a week and topic nasal bedomethasone associated with oral cetotiphen for 3 months. Since the child did not show any improvement, we indicated adenoidectoiny and placement of bilateral ventilation tubes.
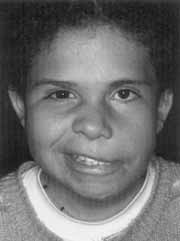
Figure 1. Goldenhar's syndrome with peripheral facial paralysis grade IV and facial asymmetry (CASE 1).
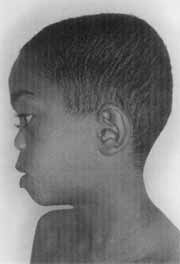
Figure 2. Large base nose and flat face, typical of Stickler's syndrome (CASE 2).
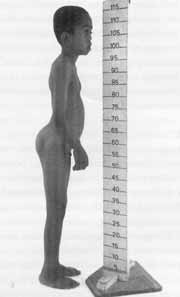
Figure 3. flat face, low height, micropenis and :artropathy are characteristics of Stickler's syndrome (CASE 2).
Case 2 - Stickler's syndrome
E. F. D., black male 9-year-old patient, from Pontal / SP, was initially treated by tile Service of Pediatric Surgery because of cryptorchidia and micropenis, and later he was referred to the Sector of Human Genetics, where the definite diagnosis of Stickler's syndrome was made. We were asked to conduct complete EST exam to confirm hypothesis of bilateral hearing loss. The mother reported that the child did not hear well, put up the TV, was dispersed and irritated. Site did not refer repetitive otitis, mouth breathing or snoring. He had delay in language development, and produced only some unintelligible sounds. Parents did not have consanguinity nor malformations in the family. The otoscopic exam revealed the presence of intact tympanic menbranes, some opacification, no retraction, normal auditory canal and low implantation of cars. He had large base nose, centered septum and normal turbinate. Oroscopy showed grade II palatine tonsils and poorly-implanted teeth. Orthopedic exam showed cracking shoulders at mobilization with salient acroimion, cracking elbows and limited extension, hips with hypermobility at rotation, cracking knees, especially on the right, and ligament increased laxity. Radiological exam revealed epiphysial dysplasia of hips, elbows and shoulders, increase of scapular-humeral articular space and bone age compatible with four years, according to G. Pyle method. He was evaluated by Ophalmology with the diagnosis of hypertnetropia, discreet ocular proptosis and hypertelorism. Pure tone audiometry showed bilateral deafness, and type C tympanometric curve with absent stapedial reflex. CT scan of temporal bones revealed bilateral Mondini's malformation of inner car, cochlea with single cavity and hypoplasic vestibule, hypoplasia of middle car with deformity and thickening of stapes and incus bilaterally. Based on the diagnosis, lie was referred to the department of Speech and Hearing Pathology to proceed with hearing aid fitting.
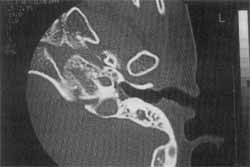
Figure 4. CT scan of left temporal bone showing Mondini's malformation in patient with Stickler's syndrome (CASE 2).
Figure 5. Spondiloeiphysial dysplasia characteristic of Stickler's syndrome (CASE 2).
Case 3 - Marshall's syndrome
R. V. D., Caucasian male 4-year-old patient, from Mirassol /SP. The mother referred a difficult pregnancy because she used hormones (sic) to force an abortion. The delivery was c-section at term. Parents did not have consanguinity and there were no cases of malformation in the family. She did not believe that the child had hearing loss or delay in development of language. He was initially seen at the service of Pediatric Neurology because of suspicion of macrocephalia. The exam revealed megalocomea, epicanthic fold, hypertelorism, discreet global hypotonia and preserved muscle strength. Ophthalmologic exam showed megalocornea and high grade myopia (14.5 diopters on the right and 16 on the left). He was referred to the Sector of Human Genetics and the diagnosis of Marshall's syndrome was confirmed. The physical exam revealed marked medial face hypoplasia, low nasal root and reduced palpebral opening. Head x-ray showed normal texture bone, craniofacial disproportion, no intracranial pathological calcifications. ENT exam did not find any other alterations; conditioned pure tone audiometry showed hearing within normal range, type A tympanometric curve and reflexes; the patient was asked to return after 6 months for audiometric follow-up.
COMMENTS
There are innumerous types of genetic hearing losses and the probability that two people will have the same genetic problem is very low. Marriage of people who have hearing disorders do not necessarily contribute to a higher incidence of hereditary hearing loss, because it is highly unlikely that these two individuals have the same genetic defect. Among the cases presented here, none of them had positive family history of congenital diseases or consanguinity among parents, confirming our statement. However, in some communities in which consanguineous marriage is frequent, the incidence of genetic hearing loss, as well as of other hereditary diseases, is much higher7.
In all cases, ENT assessment was ordered to exclude hearing loss, reinforcing the importance of the Otorhinolaryngologist for patients with genetic syndromes. In the patient with Goldenhar's syndrome (Case 1), the parents did not believe that the child had hearing loss but we confirmed mixed moderate to severe loss on the left and mild conductive loss on the right, showing the importance of good audiometric assessment, even when the complaint is not confirmed. Despite the fact that the CT scan excluded internal ear malformation, we believe that there could be a malformation of membranous labyrinth. Nevertheless, this confirmation could only be obtained by histopathology, which was not feasible7.
As to the patient with Stickler's syndrome (Case 2), the CT scan finding of Mondini's malformation of the internal ear causing bilateral sensorineural hearing loss is similar to the report by Stickler (1976), and the only alternative left is to fit hearing aids to minimize the effects on the child, even knowing that fitting may be difficult in cases like that.
In the patient with Marshall's syndrome (Case 3), we did not detect a hearing loss and pure tone audiometry and impedanciometry were within the normal range. The literature warns that in cases of progressive hearing loss 1, 4, the Otorhinolaryngologist is expected to follow up the patient with periodical exams every 6 months. The other findings of this patient were similar to those reported by the literature 1,2,3,4,6,8.
Genetic syndromes are rare entities that may affect the auditory system and the Otorhinolaryngologist is the professional expected to conduct careful assessments. Therefore, we should be able to identify and diagnose otorhinolaryngologyical pathologies that are associated with multi-systemic diseases. When we know the cause of a congenital hearing loss we better understand the hearing system and we are able to provide the best therapeutic approach to the affected patient.
REFERENCES
1. BUYSE, M. L.- Birth Defects Encyclopedia- A Service of the Center for Birth Defects Information Service, Inc., 1990, p.192193; p.504-505.
2. DALLAPICCOLA, B; MINGARELLI, R.; GENNARELLI, M; NOVELLI. G.- Genetic aspects of deafness. Acta Otorhinolaryngol-Ital., 16(2):79-80, 1996.
3. ECKEL, H.E.; RICHLING, F.; STREPPEL, M.; BOTH, B. WALGER, M.; ZOROWKA, P.- Etiology of moderate an( profound deafness in childhood. HNO, 46(3): 252-263 1998.
4. GOBLIN, R. J.; TORIELLO, H. V.; COHEN JR., M. M.- Hereditary Hearing Loss and Its Syndromes, Oxford Universit Press, New York, 1995, p.69-72, p. 120, p.193-194.
5. HOUSE, J.W.- Facial nerve grading systems. Laryngoscope 93: , 1983; pg. 1053-69.
6. MOCELLIN, M.; CAPASSO, R.; CATANI, G. S. A.; GASPERIN A. C.; VIZZOTTO JR., A. O.- Sindrome de Goldenhar (Displask Oculoauriculovertebral). Relato de caso a revisão de literatura Rev. Bras. de Otorrinolaringol., 64(1): 77-79, 1998.
7. PAPARELLA, M. M.; SCHACHERN, P. A.- Hipoacusia neurossensorial genética en niños. In: Paparella, M.M; Shumrick D. A.; Gluckman, J. L.; Meyerhoff, W. L.- .Enfermidades del oido, 3ª edição, 2: 1994 pg 1853-73
8. SMITH, S.D.- Overview of genetic auditory syndromes- J Am. Acad. Audiol., 6(1): 1-14, 1995.
9. STICKLER, G. B. E COLS.- Hereditary progressive arthro ophthalmopathy. Mayo Clin. Proc. 40: 433-455, 1965.
10. STICKLER, G. B., PUGH, D. G. - Hereditary progressive arthr-otophthalmopathy: additional observations on vertebra: abnormalities, a hearing defect, and a report of a similar case Mayo Clin. Proc. 42: 495-500, 1967.
* Resident Physician in Otorhinolaryngology at Hospital das Clínicas, Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo.
** Ph.D., Professor of the Discipline of Otorhinolaryngology at the Department of Ophthalmology and Otorhinolaryngology at Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo.
*** Associate Professor of the Department of Genetics and Biology-Applied Mathematics at Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo.
Affiliation: Department of Ophthalmology and Otorhinolaryngology at Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, and Department of Genetics and Biology-Applied Mathematics at Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo.
Study presented at I Congresso Triológico de Otorrinolaringologia, November 13 - 18, 1999, in São Paulo /SP.
Address for correspondence: Departamento de Oftalmologia a Otorrinolaringologia da Faculdade de Medicina de Ribeirão Preto, da Universidade de São Paulo.
Avenida Bandeirantes, 3.900 - 14049-900 Ribeirão Preto /SP - Tel: (55 16) 633-1586.
Article submitted on January 26, 2000. Article accepted on May 11, 2000.
Print: ![]()