Relato de Caso
Síndrome de Treacher Collins : evolução de um caso
Treacher Collins syndrome : a case evoluation
Autores:
Otavio Marambaia (Coordenador do estágio de Otorrinolaringologia do INOOA
Professor da Disciplina de Otorrinolaringologia da Escola Bahiana de Medicina e Saúde Pública -EBMSP
Chefe do corpo clínico do Hospital Santa Izabel - Santa Casa de Misericórdia de Salvador - Bahia
) Coordenador do estágio de Otorrinolaringologia do INOOA
Amaury de Machado Gomes (Especialista em ORL pela SBORL) Preceptor de Otorrinolaringologia do INOOA
Epifanio Pereira Filho (Especialista em ORL pela SBORL) Preceptor do serviço de Otorrinolaringologia do INOOA
Pablo Pinillos Marambaia (Especialista em ORL pela SBORL) Médico assistente do serviço de Otorrinolaringologia do INOOA
Ticiana Rocha Francisco (Estagiária do 2 º ano de Otorrinolaringologia do Inooa) Estagiária do 2 º ano de Otorrinolaringologia do Inooa
Palavras-Chave
Síndrome de Treacher Collins , malformações faciais congênitas
Resumo
Keywords
Treacher Collins syndrome , congenital facial malformations
Abstract
Instituição: INOOA - Instituto de Otorrinolaringologia Otorrinos Associados
Suporte Financeiro:
Introdução
As malformações congênitas da face podem levar a caracterização de síndromes específicas como a sindrome de Treacher Collins . O objetivo desse trabalho e relatar uma caso dessa síndrome e sua evolução.
Apresentação do caso
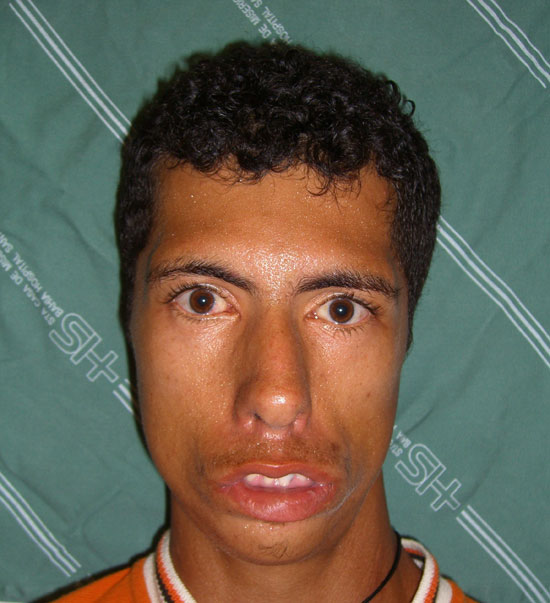
Trata-se de JPRF, 31anos, masculino, trabalhador rural, natural e procedente de Riacho da Guia, BA, trazido ao ambulatório de otorrinolaringologia por malformação crânio-facial congênita. É o quarto de seis filhos sendo que nenhum dos outros possue qualquer alteração semelhante. Não há relato de consaguinidade, uso de chás, medicamentos taratogênicos ou infecções durante a gravidez . JPRF tem boa convivência com os irmãos , entende o que é dito, apesar de não verbalizar. Interage com os familiares porém tem dificuldades de convívio social. Ao exame físico nota-se fácies atípica, sindrômica , com fissuras palpebrais inclinadas infero-lateralmente, ossos malares achatados, nariz aparentemente grande, com narinas estreitas, queixo pequeno, micrognatia, palato arqueado e microtia bilateral (Figura 1).
1 a 50.000 nascimentos 4 , e é igual nos dois sexos 1 : 1 1 . Esses pacientes apresentam fissuras palpebrais inclinadas
infero-lateralmente demonstrando uma obliquidade antimongolóide. Pode existir coloboma de íris , no terço externo da palpebra inferior (75% dos casos) e ausência dos orifícios dos condutos lacrimais inferiores. As orelhas demonstram um pavilhão auricular deformado, implantação baixa e excesso de pregueamento. Microtia pode acontecer. O conduto auditivo externo apresenta-se estreitado e agenesia acontece em até 30 % dos casos 1,2 . Defeitos nos ossículos podem
ocorrer levando a surdez de condução. Podem ocorrer fixação do martelo, ausência deste ou completa do ouvido
médio e do recesso epitimpânico. No caso descrito o paciente apresentava fissuras palpebrais inclinadas infero-lateralmente e microtia bilateral. Foi realizado estudo tomográfico que demonstrou preservação da cadeia ossicular. Já o nariz parece grande por falta do desenvolvimento do osso malar. As narinas são estreitas e as cartilagens alares hipoplásicas. A ponte nasal é alta e não se consegue detectar de forma nítida o ângulo naso-frontal . As mastóides são pneumatizadas e podem ser esclerosadas. Os seios paranasais são muito pequenos ou ausentes. A maxila é defeituosa, podendo apresentar um palato alto ou estreito. A mandíbula é hipoplásica resultando em micrognatia . Os dentes são hipoplásicos e mal posicionados . Atraso mental é observado em 5 % dos afetados 1,2,5.
O diagnóstico é clínico e baseado nessas alterações descritas. O diagnóstico diferencial deve ser feito com Síndrome de Nager ou disostose crânio-facial que é de herança recessiva e afeta as extremidades superiores além da face ; com a Síndrome de Goldenhar ou síndrome óculo-auricolo-vertebral onde os portadores apresentam microssomia facial e anomalias vertebrais.
Tratamento
As deformidades faciais podem ser melhoradas com cirurgia plástica, associado ou não com tratamento ortodôntico. Atraso mental geramente é por déficit auditivo, dessa forma, a descoberta precoce do problema permite um melhor desenvolvimento dos portadores da síndrome 3,6.
Conclusão
O desenvolvimento da genética e o grande avanço das pesquisas nessa área proporcionam o conhecimento dessas síndromes e suas principais características, permitindo assim que se promova um melhor acompanhemento dos pacientes afetados. Alguns apresentam atraso mental por falta de estimulação precoce e adaptação auditiva. Associa-se a este condições socioeconômicas precárias e falta de acesso ao profissional especializado resultando em paciente adulto, sindrômico, com déficit cognitivo e dificuldade de adaptação social. Caso o mesmo estivesse um acompanhamento específico desde o nascimento, certamente a evolução do quadro seria mais favorável.
Referências bibliográficas :
1- Oliveira AC . Malformações congênitas da face : uma revisão das sindromes mais importantes . RBORL ,Julho -Set 1982 ,vol 48 (3).
2- Gonzalez CH . Síndrome de Treacher Collins ou Síndrome de Franceschetti-Klein ou Disostose Mandíbulo-Facial.
3- Marszalek B, Wojcicki P, Kobus K, Trzeciak WH. Clinical features, treatment and genetic background of Treacher Collins syndrome. 2002. J. Appl. Genet. 43 (2) .
4- GORLIN, R.J. PINDBORG, JJ. & COHEN, M.M.JR. - Mandibulofacial Dysostosis. In GORLIN, R. J.; PINDBORG,JJ. & COHEN, M.M.JR., eds. - Syndromes of the Head and Neck. New York, Me Graw-Hill, 1976.
5-Marsh KL, Dixon MJ. Treacher Collins syndrome. Adv. Otorhinolaryngol. 2000; 56:53-9.
6-Posnick JC. Treacher Collins syndrome : perspectives in evaluation and treatment. J Oral Maxillofac Surg . 1997 Oct;55(10) : 1120-33