Artigo publicado no Caderno de Debates da RBORL:
Vol.71 ed.3 de Maio - Junho em 2005 (da página 24 à 28) |
 |
| Autor: Raquel Mezzalira1, Fernanda G. Avila2, Juliana M.A.C. Bertoncello2, João E. G. Maestri2, Eduardo Takemotto2, Oscar Maudonnet3 |
|
 |
| Relato de Caso |
| Síndrome de Waardenburg: Relato de 7 casos |
|
|
INTRODUÇÃO
A Síndrome de Waardenburg é uma doença autossômica dominante, de etiologia desconhecida, caracterizada por perda auditiva de grau variável, associada a anomalias na pigmentação dos olhos, da pele e dos cabelos, além de defeitos nos tecidos derivados da crista neural. Nem todos os pacientes com a SW apresentam todas as manifestações clínicas da doença, encontrando-se comumente as formas incompletas. Foi primeiramente descrita por um oftalmologista alemão, Petrus Johannes Waardenburg, em 1951, ao constatar que pacientes com heterocromia de íris tinham freqüentemente problemas auditivos. A incidência varia de 1 a 2 por 20.000 habitantes, não havendo predominância entre os sexos ou raças.1,2
Uma vez que a SW é herdada de forma dominante, tipicamente encontram-se famílias com várias gerações de membros que manifestam um ou mais padrões da síndrome. Muitos desconhecem possuir o gen porque sua penetrância é muito leve. De fato, encontram-se pacientes portadores do gen, porém sem manifestar característica clínica alguma, fator este que dificulta o diagnóstico clínico. A chance de transmitir o gen para os filhos é de 50% a despeito das características manifestadas.3
A SW pode ser muito mais comum do que antes se imaginava. Existem relatos que 2 a 3 crianças para cada cem que freqüentam escolas para surdos-mudos possam ser portadoras da síndrome, o que corresponderia a 2,3% da população surda congênita.4 No entanto, a maioria daqueles que portam o gen não tem perda auditiva e por conseguinte, acredita-se que para cada criança com SW e surdez severa, existam 4 a 5 membros na família com audição relativamente normal.5
A SW é classificada em 4 subtipos, determinados pelas características físicas que o paciente poderá apresentar, encontrando-se ainda classificações que englobam apenas os tipos I e II.
O tipo I é caracterizado pela evidência de telecanto associado a toda a sintomatologia da doença. Estes pacientes também possuem a ponte nasal alargada, marcada hipoplasia dos ossos nasais e maxila encurtada e retroposicionada. No tipo II o telecanto está ausente, e a surdez sensorioneural (77%) associada à heterocromia de íris (47%) são os dois indicadores diagnósticos mais importantes. O tipo III (Klein - Waardenburg) é similar ao tipo I, mas também se caracteriza por anomalias musculoesqueléticas. No tipo IV (Shah - Waardenburg) encontramos associação com o megacólon agangliônico congênito.
A Tabela 1 mostra a classificação clínica e os principais achados em cada subtipo.
O defeito genético caracteriza-se pelo não-pareamento do gen 3 levando a uma deficiência na migração das células da crista neural e conseqüentes manifestações clínicas.
Foram identificados e localizados quatro genes diferentes para a SW: PAX3, MITF, EDNRB e EDN3. Os tipos I e III têm sido associados a mutações nos genes PAX3, o tipo II ao gen MITF e o tipo IV aos genes EDNRB, EDN3 e SOX10.
A Tabela 2 traz a classificação molecular.
O gen PAX 3 localiza-se no cromossomo 2 e controla alguns aspectos do desenvolvimento da face e orelha interna. O MITF é encontrado no cromossomo 3 que também controla o desenvolvimento do ouvido e da audição.
Diante de um paciente com suspeita de SW procede-se investigação com estudos de imagem para membros superiores, avaliação oftalmológica para defeitos oculares e avaliação otorrinolaringológica para acuidade auditiva. Como os sintomas vestibulares podem ser a manifestação inicial da SW, a eletrococleografia e os testes vestibulares podem ser úteis na avaliação inicial. É importante o estudo audiológico, necessário para determinar o tipo e a severidade da disacusia. Este estudo inclui história familiar, timpanometria e audiometria de tons puros.
Os defeitos de pigmentação da pele, cabelos e olhos são similares àqueles encontrados nos portadores de albinismo parcial (piebaldismo), portanto, esta patologia deve ser considerada no diagnóstico diferencial dos pacientes com defeitos de pigmentação.
O tratamento consiste em oferecer suporte e avaliações periódicas da acuidade auditiva e ocular, além do aconselhamento genético. A reabilitação aural deve ser o primeiro método usado. Testes de linguagem e articulação podem ser necessários para intervir em erros compensatórios da articulação das palavras pela dismorfia facial.
Como prevenção, destacam-se o aconselhamento genético e a identificação precoce, que proporcionam intervenção a tempo de prevenir erros de linguagem e, no caso daqueles pacientes cofóticos, reabilitação ainda em fase de aquisição da linguagem.
DESCRIÇÃO DOS CASOS
Os autores descrevem uma família na qual a avó, 2 filhas e 4 netas apresentam recorrência familial de deficiência auditiva, heterocromia de íris e/ou poliose. Todas as pacientes foram submetidas à avaliação otorrinolaringológica, oftalmológica, audiológica e otoneurológica; e as pacientes 4 e 5, à avaliação genética que confirmou a hipótese de Síndrome de Waardenburg.
Todas as pacientes pertencem ao sexo feminino, com idades variando entre 7 e 52 anos. Como manifestações clínicas, todas apresentam hipoacusia sensorioneural que varia de grau moderado a cofose e que foi o motivo que levantou a suspeita da SW. Isocromia de íris e poliose foram encontrados em 2 pacientes e heterocromia de íris em apenas uma. Todas foram classificadas como SW tipo II conforme os achados clínicos. Com relação ao exame otoneurológico, as pacientes 4 e 6 apresentaram arreflexia calórica bilateral, e as demais achados inespecíficos como preponderância direcional.
A Tabela 3 mostra a descrição dos sete casos apresentados e os Gráficos 1 e 2 exemplificam as alterações audiométricas encontradas nas pacientes 6 e 7.
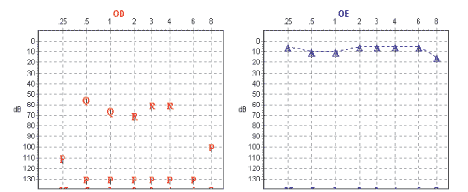 Gráfico 1. Gráfico audiométrico da paciente 7 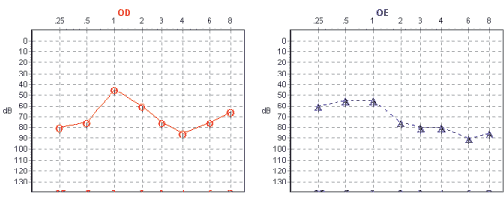 Gráfico 2. Gráfico audiométrico da paciente 6
DISCUSSÃO
O gen da SW tem três principais manifestações: audição; pigmentação da pele, olhos e cabelos; e alterações na estrutura facial, podendo os pacientes apresentarem alguns ou todos os traços da síndrome.6,7
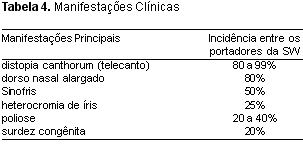
A Tabela 4 descreve a incidência das principais manifestações clínicas entre os portadores.
Os graus de perda auditiva relacionados à SW podem variar de moderados a profundos e não são progressivos. A incidência de hipoacusia na SW é relatada entre 9% a 62,5%.8 Fisch classificou os achados audiométricos em 2 padrões distintos: surdez sensorioneural total, com audição residual nas freqüências baixas e surdez sensorioneural moderada nas baixas e médias freqüências com melhora dos limiares em agudos9. Nas pacientes estudadas, a perda auditiva variou de hipoacusia sensorioneural moderada a cofose.
Existe uma grande variação nos achados audiométricos entre os pacientes sindrômicos, dentre os quais pelo menos metade não revela problemas auditivos e somente um quinto apresenta perda auditiva suficiente para influir na comunicação verbal. A surdez pode manifestar-se uni ou bilateralmente, sem necessariamente apresentar-se simétrica.10,11
O exame histopatológico da orelha interna dos pacientes com a SW tem mostrado ausência dos órgãos de Corti, atrofia do gânglio espiral da cóclea e redução nas fibras nervosas.12
Com relação à pigmentação, uma das alterações descritas é a heterocromia de íris. Usualmente um olho é azul e o outro castanho. Ocasionalmente, um misto de azul e castanho pode ser visto no mesmo olho. Está descrita na literatura uma coloração não-usual em relação aos azuis, os quais apresentam-se "muito brilhantes" (isocromia), e pode ser encontrada também pigmentação moteada perifericamente na retina (fundo albinóide). Duas das pacientes apresentam isocromia de íris e uma delas também heterocromia. Porém cabe ressaltar que nem todos os pacientes que possuem o gen têm olhos com colorações diferentes. Além disso, as manifestações oftalmológicas podem estar presentes em apenas um ou dois indivíduos de uma mesma família, característica esta que leva à suspeita em pacientes com tal alteração.
Coloração distinta no cabelo também é relatada. Mais comumente encontra-se uma mecha de fios brancos na fronte (poliose), estendendo-se para trás, que costuma estar presente em lactentes, eventualmente desaparece na infância, para somente ressurgir durante a puberdade. Dentre os casos relatados duas pacientes apresentam poliose, achado este que só foi encontrado após o seu questionamento já que as pacientes utilizam pintar o cabelo para fins estéticos.
Além disso, os portadores do gen podem ter o clareamento precoce dos cabelos relatados a partir dos 7 anos de vida.13,14
Alterações na estrutura facial podem ocorrer. O nariz costuma ser pequeno e dismórfico. As crianças freqüentemente são descritas como respiradoras bucais. Alargamento do dorso nasal simula o aumento da distância interocular, deslocando lateralmente o canto medial e o ponto lacrimal inferior (dystopia canthorum). Ainda encontram-se implantação baixa dos cabelos na linha frontal e as sobrancelhas podem juntar-se na linha média (sinofris). Áreas de vitiligo em braços e face podem ser encontradas em 15% dos casos (leucodermia).5 Nenhuma das pacientes relatadas apresenta dismorfia facial.
Em adição às três características principais, estão descritas também a associação de lábio leporino, fenda palatina (em 5% dos casos), blefarofimose, problemas colônicos como Doença de Hirschprung, elevação da escápula, defeitos nas raízes dos membros superiores, hipoplasias, defeitos na fusão carpal, sindactilias, artromiodisplasia do grande peitoral, dismorfias da coluna espinhal e estigmas da Síndrome de Melkerson-Rosenthal.1,14,15 Tais características não foram encontradas nas pacientes estudadas.
Sintomas vestibulares também foram relatados. Black et al. estudaram 50 pacientes portadores da SW que apresentavam sintomas vestibulares compatíveis com hidropsia endolinfática.13 A anomalia mais consistente foi uma elevação dos segmentos SP/AP na eletrococleografia. Tal fato foi observado em 80% dos pacientes, dentre os quais somente 10% apresentavam perda auditiva. Todos os pacientes, exceto àqueles com hipoacusia, tinham respostas auditivas centrais normais. Os testes de função vestíbulo-ocular e vestíbulo-espinhal foram anormais na maioria, mas sem demonstrar padrão específico. Encontramos arreflexia vestibular em duas pacientes e nas demais achados inespecíficos como preponderância direcional. Interessante observar que nenhuma apresentou exame otoneurológico normal.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Reed WB, Stone VM, Boder E, Ziprkowski L. Pigmentary disorders in association with congenital deafness. Arch Dermatol 1967; 95: 176-86.
2. Ortonne J-P, Mosher DB, Fitzpatrick TB. Waardenburg Syndrome. In: Vitiligo and Other Hypomelanoses of Hair and Skin New York: Plenum Medical Book Co 1983; 337-68.
3. Shprintzen R. Deafness and Other Communication Disorderstiology and Prevention of Communicative Disorders. San Diego: Singular Publishing Group Inc 1998; 147-99.
4. Gilbert P. A-Z Reference Book of Syndromes and Inherited Disorders 1st Ed. London: Chapman and Hall; 1996. p. 332-5.
5. National Center for Biotechnology Information. National Library of Medicine, National Institutes of Health Waardenburg Syndrome Online US West 11 Oct 1999.
6. National Institute on Deafness and Other Communication Disorders. Waardenburg Syndrome Online US West19 Oct 1999.
7. Waardenburg PJ. A new syndrome combining developmental anomalies of the eyelids, eyebrows, and nose root with pigmentary defects of the iris and head hair and with congenital deafness. IS J Hum Genet 1951; 3: 195-253.
8. Susi Tsafir J. Light-Eyed Negroes and the Klein-Waardenburg Syndrome London: Macmillan, 1974.
9. Fish L. Deafness as part of an hereditary syndrome. J Laryngol Otol 1959; 73: 353-62.
10. Read A, Newton VE. Waardenburg Syndrome. Journal of Medical Genet 1997; 34:656-65.
11. Read A, Newton VE. Waardenburg Syndrome Genetics and Hearing Impairments. San Diego: Singular Publishing Group Inc, 1996.
12. Friedman I, Fish L. Deafness as part of an hereditary syndrome. J Laryngol Otol 1959; 73: 363-82.
13. Shprintzen R. Genetics, Syndromes and Communication Disorders. San Diego: Singular Publishing Group Inc, 1997.
14. Hedge MN. Pocket Guide to Treatment in Speech-Language Pathology. 1st Ed, San Diego, Singular Publishing Group Inc, 1996; 130-1.
15. Dourmishev AL, Dourmishev LA, Schwartz RA, Janniger CK. Waardenburg Syndrome. Int J Dermatol 1999; 38: 656-63.
1 Médica do Instituto Penido Burnier.
2 Médicos Residentes do Instituto de Otorrinolaringologia Penido Burnier.
3 Médico Otoneurologista.
Clínica de Otorrinolaringologia do Instituto Penido Burnier, Campinas, SP.
Trabalho apresentado como pôster no II Congresso Triológico de Otorrinolaringologia em Goiânia - 2001.
Endereço para correspondência: Fernanda Gobbi de Avila - Av. Andrade Neves 611 Campinas SP 13013-161.
Tel (0xx19) 3236-1027 - E-mail: fgavila@bol.com.br
Artigo recebido em 03 de outubro de 2003. Artigo aceito em 15 de janeiro de 2004.
|
|
|
|
Todos os direitos reservados © Associação Brasileira de Otorrinolaringologia e Cirurgia Cérvico Facial
Av. Indianópolis, 740 - Moema - São Paulo - SP - Brasil - Fone: (11) 5052.9515 |
|
 |
|