

Caderno de Debates (Suplementos)
![]() Bem vindo ao nosso Caderno de Debates!
Bem vindo ao nosso Caderno de Debates!
Artigo publicado no Caderno de Debates da RBORL:
Vol.71 ed.6 de Novembro - Dezembro em 2005 (da página 7 à 11)
Autor: Willer Moreira Costa Júnior1, Marcos José Burle Aguiar2, Helena Maria Gonçalves Becker3, Mônica Maria de Oliveira Melo4, Letícia Paiva Franco5, Marcelo de Moura6
Relato de Caso
Manifestações otorrinolaringológicas na síndrome de Pfeiffer: revisão de literatura e relato de um caso da síndrome - Pfeiffer tipo I
![]()
Introdução
A Síndrome de Pfeiffer é uma doença com padrão de transmissão autossômica dominante que consiste em craniossinostose, hipoplasia médio-facial, nariz pequeno com ponte nasal baixa, prognatismo relativo, hipertelorismo associado a alterações de membros como polegares largos e desviados radialmente e háluces largos e grandes, dentre outras anomalias1,2.
A Síndrome de Pfeiffer foi por muito tempo relacionada à Síndrome de Apert, porém com expressão mais branda, ou alternativamente com a Síndrome de Crouzon, porém com superexpressão das anomalias em membros. Entretanto, todos os estudos falharam em demonstrar transições de uma síndrome para outra nos diversos fenótipos de famílias estudadas, o que confirma a Síndrome de Pfeiffer como uma entidade distinta no grupo das acrocefalossindactilias3.
Essa síndrome pode ser dividida em três subtipos com manifestações e prognósticos distintos, ainda que não possam ser classificados como entidades geneticamente e nosologicamente separadas, pela sobreposição de características clínicas1,2,4. Em todos os três subtipos da Síndrome de Pfeiffer as manifestações otorrinolaringológicas são importantes e comuns, sendo o objetivo do presente estudo descrevê-las.
Para ilustrar esse trabalho, descrevemos um caso da síndrome de Pfeiffer Tipo I atendido no Hospital das Clínicas da Faculdade de Medicina da Universidade Federal de Minas Gerais.
Apresentação do Caso
ESC, 2 meses de idade, quarta filha de um casal da raça branca não consangüíneo. Sua gravidez transcorreu sem intercorrências e não houve relato de exposição a teratógenos. Parto ocorreu na 39ª semana de gravidez de maneira espontânea por via vaginal. Índice de Apgar foi de 7 no 1º minuto e 9 no 10º minuto. A criança recebeu alta hospitalar no segundo dia de internação sendo encaminhada ao Serviço de Genética para avaliação.
Exame físico realizado no segundo mês de vida: Peso 2730g (percentil 25), comprimento 47cm (percentil 10) e perímetro cefálico 47cm (percentil 25). Apresentava fáscies grosseira, craniossinostose associada a turribraquicefalia, hipoplasia médio-facial com proptose ocular bilateral moderada, pregas epicantais, fendas palpebrais discretamente inclinadas para baixo, nariz com base alargada e ponte nasal baixa. O palato duro era moderadamente ogival. Observava-se ainda conduto auditivo externo atrésico no terço lateral, com porção medial terminando em fundo de saco bilateralmente. No exame dos membros destacava-se a presença de polegares alargados com abdução da falange distal e háluces varos e alargados. Foram observadas ainda prega simiesca na mão esquerda e fosseta sacral sem trajeto fistuloso, além de uma hérnia umbilical. Não apresentava evidências de comprometimento do desenvolvimento neuropsicomotor e conseguia firmar parcialmente o pescoço quando sentada. Logo a seguir foi encaminhada para realização de cariótipo em sangue periférico, cujo resultado foi 46 XX - normal.
A criança foi então encaminhada ao Serviço de Otorrinolaringologia da UFMG, onde foram realizados os exames de Fibronasofaringolaringoscopia - que demonstrou a presença de fossas nasais muito estreitadas com obstrução da região posterior à esquerda - e Audiometria de tronco cerebral (BERA), com presença de ondas I a V com amplitudes e intervalos interpicos normais com atraso da latência absoluta de todas as ondas bilateralmente e limiar em torno de 55dB. O estudo tomográfico realizado na mesma época revelou presença de atresia óssea do conduto auditivo externo medialmente em ambas as orelhas e presença de ossículos malformados fundidos à placa atrésica. Notava-se ainda fossas nasais estreitadas com atresia coanal à esquerda. À seguir, a criança foi encaminhada à neurocirurgia para avaliação e correção da craniossinostose e à fonoterapia para estimulação.
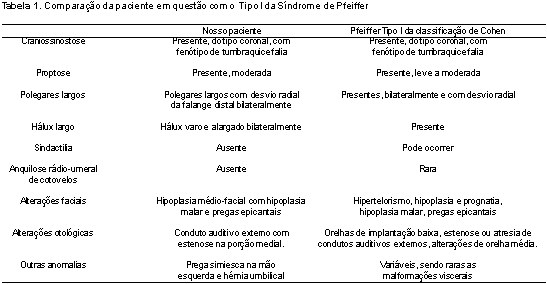
Considerando-se o padrão de malformações apresentadas pela criança, foi estabelecido o diagnóstico de Síndrome de Pfeiffer e a paciente classificada no subtipo I da classificação de Cohen, pelas semelhanças observadas entre esse subtipo e o fenótipo apresentado pela mesma, como se observa na Tabela 1.
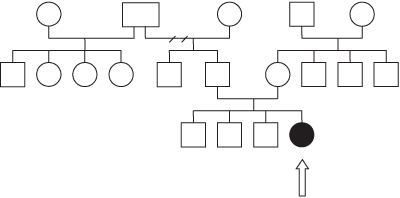
O heredograma da paciente demonstra que, nesse caso, a Síndrome de Pfeiffer Tipo I ocorreu como caso esporádico, provavelmente devido a uma nova mutação na família, como se observa na Figura 1 (Heredograma). Não obstante, sabemos que a maior parte dos casos observados do subtipo I são familiares, com padrão de transmissão autossômico dominante.
Discussão
Inicialmente descrita por Pfeiffer em 1964 e posteriormente também denominada acrocefalosindactilia tipo V (1969), a Síndrome de Pfeiffer tem um padrão de transmissão autossômico dominante com penetrância completa e expressividade variável resultante de uma mutação nos genes dos fatores de crescimento de fibroblastos (FGFR), assim como outras síndromes que envolvem craniossinostose. Tanto nas formas esporádicas quanto nas formas familiares ocorrem mutações nos genes FGFR1 e FGFR5. Rutland et al.6 sugerem que existem diferenças clínicas nos pacientes que apresentam mutações em um ou outro gene, sendo que os casos mais graves estão associados a mutações no gene FGFR2.
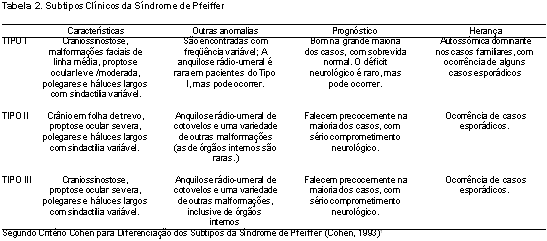
Através de estudos detalhados das manifestações clínicas, dos achados radiológicos, evolução natural da doença e da freqüência de anomalias associadas, a Síndrome de Pfeiffer foi dividida em três subtipos clínicos por Michael Cohen1, em 1993.
Na Síndrome de Pfeiffer Tipo I a maior parte dos pacientes apresentam desenvolvimento neuropsicomotor e inteligência normal ou próxima do normal. No entanto, em uma pequena parte observa-se deficiência mental leve a moderada. Suas principais características são craniossinostose, malformações faciais de linha média (incluindo retração facial, má-oclusão dentária e mordida aberta), polegares largos e desviados radialmente e háluces largos, com grau de sindactilia variável. Nesse subtipo a craniossinostose geralmente se manifesta com turribraquicefalia, pelo fechamento precoce das suturas sagital e coronal bilateralmente, com predominância do diâmetro biparietal sobre o diâmetro occiptofrontal. Também são observados hipertelorismo, proptose ocular leve, pregas epicantais, hipoplasia maxilar, prognatismo, orelhas de implantação baixa e estenose ou atresia de conduto auditivo dentre outras malformações variáveis. Esse subtipo segue o padrão autossômico dominante de transmissão, com vários casos familiares descritos - Pfeiffer (1964, 1969); Noack (1958); Degenhart (1964); Zippel and Schüler (1969); Martsolf et al. (1971) e Cracco et al. (1970), dentre outros - ainda que existam relatos de casos esporádicos, relacionados a mutações novas.
O Tipo II da Síndrome de Pfeiffer manifesta-se principalmente pela craniossinostose do tipo "cloverleaf skull" ou crânio em folha de trevo, caracterizado pelo fechamento precoce das suturas coronal, sagital e lambdóide, com protusão das duas regiões parietais e do ápice da calota craniana, dando o aspecto de trevo de três folhas ao corte coronal. Polegares largos com háluces grandes e largos e sindactilia variável também estão presentes, uma vez que fazem parte da definição da síndrome1.
Também é característica do Tipo II a proptose ocular intensa, para a qual contribui significativamente a retração facial de linha média. Uma variedade de anomalias palpebrais e do globo ocular se desenvolve em conseqüência do encurtamento orbitário anterior manifesto clinicamente por órbitas rasas associadas a fissuras palpebrais inclinadas obliquamente, retração palpebral e cicatrizes de córnea. Outro achado importante é a anquilose rádio-umeral de cotovelos, além do grave envolvimento do sistema nervoso central, manifesto caracteristicamente por hidrocefalia. Hipertelorismo, orelhas rodadas e de implantação baixa, alargamento da pirâmide nasal com encurtamento do nariz, malformações de vértebras cervicais, malformações torácicas, atresia de coanas e do conduto auditivo externo são outras alterações freqüentemente vistas nesses pacientes. Anormalidades dos órgãos internos não são comuns, exceto as anomalias cerebrais. O subtipo II é de ocorrência esporádica, assim como o subtipo III.
A principal diferença entre os subtipos II e III da Síndrome de Pfeiffer é a ausência do crânio em forma de trevo nesse último, o que dificulta o reconhecimento dos pacientes portadores do subtipo III, além da maior freqüência de anomalias viscerais. Ambos apresentam um prognóstico ruim com comprometimento neurológico grave e evolução precoce para o óbito1,2. Embora a literatura tenha descrito com detalhe as alterações otorrinolaringológicas relacionadas às síndromes de Apert e Crouzon (Bergström et al., 1972; Selder, 1973; Lindsay et al., 1975; Calderelli, 1977; Gould and Calderelli, 1982; Phillips and Miyamoto, 1986; Northern and Downs, 1991), o mesmo não ocorre em relação à Síndrome de Pfeiffer. As descrições relativas a patologias auditivas são superficiais ou limitam-se ao estudo de casos individuais, como o trabalho realizado por Cremers7, em 1981. Neste estudo é relatado o caso de um garoto de 14 anos, portador do Tipo I da Síndrome de Pfeiffer, com perda auditiva de condução leve e malformações de orelha média. Posteriormente, em 1995, Moore et al.3 estudaram um grupo de 14 pacientes portadores de variados subtipos da síndrome de Pfeiffer e demonstraram que a efusão em orelha média foi um achado universal, assim como a estenose de nasofaringe. Encontraram também déficit auditivo significativo em um paciente do Tipo III da síndrome, embora a audiometria não tenha sido realizada em nenhum dos pacientes. A Tabela 2 mostra de forma comparativa os três subtipos clínicos da Síndrome de Pfeiffer.
Vallino-Napoli8, em 1995, percebendo então a escassez desses relatos, estudou um grupo de nove pacientes, 5 do sexo feminino e 4 do sexo masculino, todos portadores da Síndrome de Pfeiffer, no intuito de estabelecer a prevalência de problemas audiológicos e otopatologias associadas à síndrome. Os resultados de seu trabalho mostraram que a perda auditiva é um achado comum associado à síndrome, sendo que dos nove pacientes estudados, cinco apresentaram perda auditiva condutiva do tipo moderada (41 a 55 dB HL) e um apresentou perda auditiva severa mista (71 a 90 dB HL). Dos três pacientes restantes, dois apresentavam perdas leves de condução e apenas um apresentava audição normal. A perda auditiva, em todos os casos, revelou-se bilateral e simétrica.
Outras anomalias comumente observadas nesses pacientes foram as malformações do conduto auditivo externo e deformidades da orelha média e ossículos. As malformações de orelha externa consistiram predominantemente de estenose ou atresia das porções óssea e/ou cartilaginosa do conduto auditivo e hipoplasia da cavidade da orelha média ou dos ossículos. Quando presentes, essas deformidades foram consideradas causas primárias das perdas auditivas observadas.
As anomalias de orelha média, ossículos, membrana timpânica, conduto auditivo externo, maxila e mandíbula são originadas de distúrbios do desenvolvimento dos arcos branquiais, particularmente do primeiro e segundo arcos. Falhas de desenvolvimento do primeiro arco branquial também podem afetar negativamente as funções da tuba auditiva e ainda a pneumatização das células aeradas da mastóide. Assim, as efusões de orelha média são uma conseqüência natural dessas anormalidades anatômicas e disfunções mecânicas, sendo a otoscopia um exame de imensurável valor nesses casos e que, infelizmente, pode estar impedido pela estenose ou atresia de conduto.
A obstrução de vias aéreas nesses pacientes é resultado da obstrução das vias aéreas superior e inferior. Nos primeiros meses de vida um estreitamento grave das vias aéreas superiores pode levar à morte, como ocorre principalmente nos pacientes do subtipo II. Esses casos onde previamente os pacientes eram submetidos diretamente à traqueostomia estão sendo submetidos inicialmente a um procedimento de alargamento da nasofaringe por ressecção de partes moles quando possível. Nos casos onde ocorre uma obstrução moderada, o ambiente de hipoxemia crônica dificulta o controle da pressão intracraniana podendo comprometer o desenvolvimento neurológico3. Nesses pacientes também a ressecção de partes moles associada ou não a adenoamigdalectomia pode ser utilizada. Naqueles casos refratários ou recorrentes ao tratamento inicial os avanços médio-faciais podem ser realizados em um segundo tempo quando já existe uma maturação dessa porção facial3.
Outras anomalias craniofaciais incluem assimetrias faciais, hipoplasia maxilar com prognatismo relativo o que contribui para o baixo desenvolvimento da região médio-facial levando em última análise a um estreitamento das fossas nasais e nasofaringe associando, por vezes, a atresia coanal.
No diagnóstico diferencial da síndrome de Pfeiffer devemos incluir principalmente as outras síndromes que apresentam craniossinostose como Apert, Crouzon, Saethre-Chotzen e Jackson-Weiss1.
Figura 1. Heredograma da paciente em questão
Comentários Finais
A Síndrome de Pfeiffer deve ser considerada no diagnóstico diferencial das síndromes que envolvem craniossinostose com alterações médio-faciais e de arcos branquiais. O seu conhecimento nos fornece informações importantes sobre sua evolução clínica facilitando intervenções precoces quando possíveis.
Referências Bibliográficas
1. Cohen MM Jr. Pfeiffer syndrome update, clinical subtypes, and guidelines for differential diagnosis. Am J Med Genet 1993; 45: 300-7.
2. Cohen MM Jr, MacLean RE. Craniossynostosis: Diagnosis, Evaluation, and Management. 2nd ed. New York: Oxford University Press; 1999.
3. Moore MH. Pfeiffer Syndrome. A clinical review. Clef Palate Craniofac J 1995; 32: 62-70.
4. Cohen MM Jr. Favorable prognosis for children with Pfeiffer syndrome types 2 and 3: Implications for Classification. Am J Med Genet 1998; 75: 240-4.
5. Say B, Can B, Anderson C, Schaefer F. Novel mutation in the FGFR2 gene at the same codon as the Crouzon syndrome mutations in a severe Pfeiffer syndrome type 2 case. Am J Med Genet 1998; 75: 252-5
6. Rutland R et al. Identical mutations in the FGFR2 gene cause both Pfeiffer and Crouzon syndrome phenotypes. Nature Genet 1995; 9:173-6.
7. Cremers CWRJ. Hearing loss in Pfeiffer's syndrome. Int J Pediatr Otorhinolaryngol 1981; 3: 343-53.
8. Vallino Napoli LD. Audiologic and otologic characteristics of Pfeiffer syndrome. Clef Palate Craniofacial J 1996; 33: 524-9.
1 Médico Residente em Otorrinolaringologia pelo Hospital das Clínicas - UFMG.
2 Professor Adjunto do Departamento de Pediatria da Faculdade de Medicina da Universidade Federal de Minas Gerais, Especialista em Genética Médica e Coordenador do Serviço Especial de Genética Médica do Hospital das Clínicas - UFMG.
3 Professora Adjunta do Departamento de Otorrinolaringologia, Oftalmologia e Fonoaudiologia da Faculdade de Medicina - UFMG.
4 Acadêmica de Medicina pela Faculdade de Medicina da Universidade Federal de Minas Gerais.
5 Médica Otorrinolaringologista Mestranda em Otorrinolaringologia pelo Hospital das Clínicas - UFMG.
6 Médico Residente em Otorrinolaringologia pelo Hospital das Clínicas - UFMG.
Instituição: Departamento de Otorrinolaringologia, Oftalmologia e Fonoaudiologia da Universidade Federal de Minas Gerais.
Endereço para correspondência: Dra. Helena Maria Gonçalves Becker - Avenida Pasteur 88 4º andar Bairro Santa Efigênia Belo Horizonte MG 30150-290.
Tel./Fax: (0xx31) 3222-2891/ (0xx31) 99724632
Artigo recebido em 18 de maio de 2004. Artigo aceito em 17 de junho de 2004.
Versão para impressão: ![]()
All rights reserved - 1933 /
2026
© - Associação Brasileira de Otorrinolaringologia e Cirurgia Cérvico Facial