

Caderno de Debates (Suplementos)
![]() Bem vindo ao nosso Caderno de Debates!
Bem vindo ao nosso Caderno de Debates!
Artigo publicado no Caderno de Debates da RBORL:
Vol.71 ed.3 de Maio - Junho em 2005 (da página 37 à 40)
Autor: Fábio Zerati1, Sulene Pirana2, Elisama Queiroz3, Luis Ubirajara Sennes4
Relato de Caso
Ressecção parcial de língua em paciente com macroglossia portador de síndrome de Beckwith-Wiedemann: relato de caso
![]()
INTRODUÇÃO
A Síndrome de Beckwith-Wiedemann foi descrita primeiramente por Beckwith em 1963, e posteriormente por Wiedemann, em 1964, como Síndrome E.M.G. (exonfalo-onfalocele, macroglossia e gigantismo), sendo relacionada atualmente a outras malformações1,2. É uma síndrome genética de supercrescimento, relativamente comum, que se caracteriza pela presença de anomalias congênitas como visceromegalias, macroglossia, defeitos de parede abdominal, supercrescimento pré e pós-natal e hipoglicemia neonatal. Trata-se, portanto, de uma síndrome polimorfa, sujeita a uma associação variável de sinais e sintomas. Dentre as inúmeras anomalias citadas, a macroglossia é a manifestação mais comum da síndrome, estando presente em 82% a 99% dos indivíduos acometidos, podendo estar associada a um espectro de alterações crânio-faciais3. Pode causar dificuldade para deglutição, fonação e até problemas respiratórios, pela incapacidade de colocar toda a língua dentro da boca. Muitos autores abordam essa anomalia de maneira clínica, aguardando o crescimento da criança, uma vez que a língua tende a acomodar-se na boca com o passar da idade. No entanto, há casos em que a resolução cirúrgica é mandatória4-7.
Relato de caso
Paciente masculino, MRMSJ, 3 anos, foi atendido no ambulatório de otorrinolaringologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo com quadro de macroglossia importante, ocasionando dificuldade de deglutição devido à impossibilidade de mastigação.
Filho de pai e mãe saudáveis, não-consangüíneos, de 25 e 21 anos, respectivamente. A mãe da criança havia tido 3 gestações, sendo que o primeiro filho havia morrido aos 8 meses de gestação devido à macrossomia, e o segundo era saudável. A gestação do paciente relatado foi sem intercorrências, não havendo uso de drogas ou infecções associadas. Nascido de parto normal com 32 semanas (houve uso de ocitocina) com 3360Kg, estatura de 48cm, Apgar no 1o minuto de 7 e no 5o minuto de 6, apresentou hipoglicemia. Foi mantido com SNG devido à dificuldade de se alimentar pela macroglossia, conseguindo mamar somente no 15o dia de vida. Apresentou icterícia neonatal, tratada com fototerapia. Observou-se hemangioma plano em glabela, nariz, nuca, dorso e abdome, e, ainda, diástase do músculo reto abdominal, hepatoesplenomegalia congênita, assimetria renal (rim esquerdo maior que o direito por cisto cortical homogêneo de aproximadamente 2,5cm de diâmetro- suspeita de nefroblastose). Aos 5 meses de vida, apresentou broncopneumonia e derrame pleural.
No atendimento ambulatorial, a mãe relatava grande dificuldade de alimentação, conseguindo deglutir apenas líquidos e pastosos, pois havia impossibilidade de mastigação devido à macroglossia. Apresentava a boca constantemente aberta, com a língua interposta entre as arcadas dentárias (Figura 1), roncos intensos à noite com episódios de apnéia, sem queixa de sintomas de rinite. Teve 3 episódios de otorréia desde o nascimento. Foi submetido à audiometria condicionada com respostas adequadas.
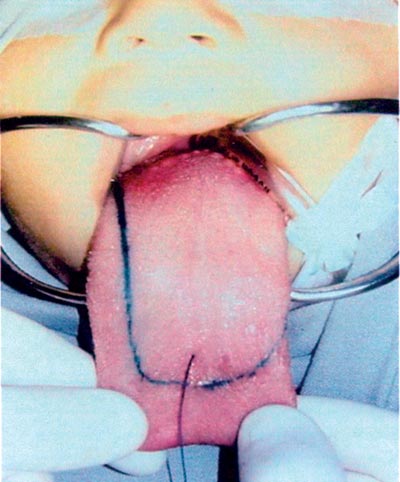
De acordo com o quadro descrito acima, optou-se em realizar tratamento cirúrgico para a macroglossia. A ressecção cirúrgica foi programada para diminuir tanto a largura quanto o comprimento da língua, pois a mesma impedia o fechamento da boca e a oclusão dentária anterior e lateralmente. A glossectomia foi realizada retirando-se parte da margem lateral direita e porção anterior da língua (Figura 2). As margens da incisão foram suturadas borda à borda com vicryl 3-0 em pontos simples separados (Figura 3). A margem direita da língua era maior que a esquerda, havendo, portanto, uma assimetria da mesma.
No pós-operatório (30o dia), o paciente apresentava boa cicatrização da língua e esta podia ser mantida totalmente dentro da boca com os lábios cerrados. A criança conseguia ingerir sólidos, mastigando os alimentos. Não apresentava mais respiração bucal e os roncos tinham cessado (Figura 4). A motilidade da língua não sofreu alteração.
Figura 1. Macroglossia em criança com Síndrome de Beckwith-Wiedemann.
Figura 2. Margens da ressecção cirúrgica (glossectomia parcial).
Figura 3. Margens suturadas com pontos simples, fio absorvível.
Figura 4. No 30º dia pós-operatório, o paciente apresentava a língua totalmente dentro da boca, com os lábios cerrados.
Discussão de caso e revisão de literatura
Uma associação entre aumento de glândulas adrenais, hemihipertrofia, macroglossia, onfalocele e hiperplasia de ilhotas pancreáticas foi descrito primeiramente por Beatty e Hawes em 1955. A Síndrome de Beckwith-Wiedemann foi reconhecida pela primeira vez como síndrome por Beckwith em 1963, quando ele descreveu achados de necropsia de 3 crianças com onfalocele, macroglossia, aumento do córtex adrenal, displasia medular renal e visceromegalias hiperplásicas. Em 1964, Wiedemann publicou um artigo sobre trigêmeos com onfalocele e filhos de pais consangüíneos. Ele descreveu também as complicações neonatais da macrossomia e da hipoglicemia. Por esse motivo, ficou conhecida como síndrome de Beckwith-Wiedemann.1,2
Trata-se de uma síndrome genética de supercrescimento, relativamente comum, que se caracteriza pela presença de anomalias congênitas, como visceromegalias, macroglossia, defeitos de parede abdominal, supercrescimento pré e pós-natal e hipoglicemia neonatal. Hérnia umbilical, macroglossia e gigantismo são considerados os achados diagnósticos característicos da síndrome, e por esse motivo, a mesma é também chamada de síndrome EMG (exomphalos, macroglossia e gigantism).
Para ser estabelecida como tal, é necessária a presença de 3 dos 4 sinais maiores, que são: patologia onfálica, macroglossia, gigantismo pré e/ou pós-natal e hipoglicemia, além de alguns sinais menores, a saber: dismorfia crânio-facial, nevus facial, globos oculares proeminentes, raiz nasal aplanada, anomalias do lóbulo da orelha, protuberância occipital, microcefalia, visceromegalias, alterações gênito-urinárias, hemihipertrofia, idade óssea avançada, atraso mental, malformações digestivas, hérnia diafragmática, policitemia, hipocalcemia.
Macroglossia é a manifestação mais comum da síndrome, estando presente em 82% a 99% dos indivíduos acometidos3. Outros estudos revelam a macroglossia estando presente em 71% a 87% dos casos, enquanto anormalidades da parede abdominal, desde hérnia umbilical até diástase do músculo reto abdominal, aparecem numa taxa que varia de 75% a 77%1,2.
Outros achados comuns incluem anormalidades renais e gênito-urinárias (47%-62%), nevus na face (32%-62%), entre outros. Hipoglicemia está presente em aproximadamente 50% dos indivíduos com a síndrome, estando relacionada com hiperinsulinemia decorrente de hiperplasia de ilhotas pancreáticas. A hipoglicemia, quando não diagnosticada, pode persistir por vários meses de vida e levar à prejuízo neurológico, que é visto em 9% a 12% dos pacientes com essa síndrome. O início dos sintomas ocorre no 2o ou 3o dia de vida, sendo persistente, severa e de difícil tratamento. A hemihipertrofia está presente em 12% a 23% dos indivíduos e relaciona-se com risco aumentado de desenvolvimento de neoplasias benignas e malignas. As neoplasias mais comuns nesses pacientes são nefroblastoma, carcinoma adrenocortical, hepatoblastoma1-3. Exame histopatológico do rim demonstra restos de nefrogênese anormal, o que justifica a incidência aumentada de nefroblastoma. O achado de que 7,5% dos pacientes com a síndrome desenvolvem neoplasias, a maioria malignas, permite a padronização do uso da ultra-sonografia abdominal como método de detecção precoce dessas lesões, além da determinação de alfa-fetoproteína sérica1.
A incidência dessa síndrome é estimada em 0,07 em 1000 nascidos vivos, sendo 15% dos casos hereditários e o restante (85%), esporádico. Anormalidades cromossômicas foram descritas envolvendo o braço curto do cromossomo 118-10. No entanto, o mecanismo preciso de modificação genética permanece obscuro até o momento, mas já se sabe que essa condição pode ocorrer de três maneiras: 1) casos familiares com associação genética do 11p15; 2) casos com anormalidades de cariótipo, devido a duplicações paternas do 11p15 ou a translocações maternas, ou ainda, a inversões com quebras no 11p15; 3) casos esporádicos. Os casos familiares são incluídos nas heranças autossômicas dominantes com penetrância incompleta. Muitos casos hereditários nascem com herança materna e a penetrância parece ser muito mais baixa quando a síndrome é relacionada ao sexo masculino.
Evidências recentes têm questionado a participação do fator de crescimento como a insulina 2 (IGF-2) na patogênese da síndrome. Estudos genéticos mapearam o gene responsável pela produção do IGF-2 no cromossomo 11p15 e observou-se que a produção dessa molécula durante o desenvolvimento fetal é maior em pacientes com a síndrome de Beckwith- Wiedemann, levando os tecidos a um supercrescimento8,10.
Macroglossia é a manifestação mais comum da síndrome, apresentando-se em vários níveis, e pode estar associada a um espectro de alterações crânio-faciais que necessitam de intervenção. A via aérea está comprometida em repouso, em caráter intermitente, relacionada à posição, ou durante à alimentação. Isso pode causar dificuldade de deglutição, de alimentação, e de manejo com a saliva. Crianças mais velhas e adultos podem ter dificuldades com a fala. A presença de uma língua aumentada faz com que o paciente tenha um aspecto de retardo mental. A incapacidade de colocar a língua dentro da boca pode acarretar ressecamento, ulcerações e até infecções da ponta da mesma. Além disso, a macroglossia não-tratada pode ocasionar um desenvolvimento crânio-facial alterado, com mordida aberta, dentes incisivos inclinados, levando a um aspecto facial prognata.3,11,12. Os achados histológicos da macroglossia mostram uma hiperplasia muscular ou até um aspecto histológico normal.
Em geral, a macroglossia nessa síndrome tende a diminuir com a idade, durante a infância e adolescência, devido ao crescimento das estruturas da cavidade oral e ao reposicionamento ântero-posterior da base da língua. Porém, mais da metade dos casos necessita de intervenção cirúrgica antes dessa fase4.
A obstrução de vias aéreas superiores em recém-nascidos com a síndrome parece estar relacionada à macroglossia, enquanto que na infância parece resultar de uma hipertrofia adenoamigdaliana. Assim, um trabalho realizado em 1995 por Rimell et al.3 recomendou traqueostomia no 1o caso de obstrução, até o crescimento permitir a acomodação da língua na cavidade oral. Com relação à dificuldade de deglutição, o mesmo autor propõe mudanças na consistência da comida para facilitar a alimentação da criança. Para a obstrução da infância, o tratamento proposto pelo trabalho seria adenoidectomia e amigdalectomia, com excelentes resultados, já que muitos pacientes permanecem com obstrução mesmo após ressecção parcial da língua. No entanto, Mixter et al.5 obtiveram melhora dos sintomas obstrutivos após glossoplastia redutora com técnica de redução central da língua, retirando em forma de w a porção posterior da língua.
Diante do que foi exposto, há uma série de indicações para o tratamento da macroglossia na síndrome de Beckwith-Wiedemann, sendo a glossectomia parcial o procedimento mais realizado nesses casos, com o objetivo de reduzir a língua ao tamanho normal e preservar sua função. Várias técnicas de glossoplastia redutora foram classificadas por Valcek et al.4,7, que as dividiu em vertical, central, marginal, pedículo ântero-lateral e horizontal. A técnica utilizada no caso relatado foi a ressecção lateral das margens da língua, sendo a margem direita maior que a esquerda, devido à assimetria existente. Observou-se um excelente resultado estético e funcional, com melhora dos roncos, da aparência, da alimentação e da fala após a cirurgia. A criança pôde desenvolver a fala e respirar adequadamente, evitando-se problemas futuros de alteração crânio-facial.
O tratamento cirúrgico da macroglossia promove um aprimoramento estético e funcional, e sendo realizado precocemente, pode prevenir complicações dento-alveolares. Há também melhora da fala, da deglutição e do manejo da saliva. Pode ocorrer, porém, complicações relacionadas à ressecção parcial da língua, como anquilose, língua globular com insensibilidade da ponta13. O corpo da língua pode permanecer largo, mesmo que o novo tamanho seja normal. Não foram observadas quaisquer complicações pós-operatórias com a técnica cirúrgica empregada nesse caso.
Conclusão
A macroglossia é uma característica marcante da Síndrome de Beckwith-Wiedemann, necessitando, em certos casos, de tratamento cirúrgico. A glossoplastia redutora tem excelentes resultados na literatura, comprovados no caso relatado, com melhoria dos sintomas obstrutivos e estéticos. Além disso, tem-se mostrado efetiva no aperfeiçoamento da aquisição da fala em indivíduos sem deficiências neurológicas centrais.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Weng EY, Mortier GR, Graham Jr JM. Beckwith-Wiedemann Syndrome. Clin Pediatr 1995; 13(5): 317.
2. Wu NFT, Kushnick T. The Beckwith-Wiedemann Syndrome. Clin Pediatr 1974; 13: 5.
3. Rimell FL, Shapiro AM, Shoemaker DL, Kenna MA. Head and neck manifestations of Beckwith-Wiedemann syndrome. Otolaryngol Head Neck Surg 1995; 113: 262-5.
4. Morgan W, Friedman EM, Duncan NO, Sulek M. Surgical Management of Macroglossia in Children. Arch Otolaryngol Head Neck Surg 1996; 122: 326-9.
5. Mixter RC, Ewanowski SJ, Carson LV. Central Tongue Reduction for Macroglossia. Plastic Reconstr Surg 1993; 91(6); 1159-62.
6. Siddiqui A, Pensler JM. The Efficacy of Tongue Resection in Treatment of Symptomatic Macroglossia in the Child. Ann Plast Surg 1990; 25: 14.
7. Kveim M, Fisher JC, Jones KL, Gruer B. Early Tongue Resection for Beckwith-Wiedemann Macroglossia. Ann Plast Surg 1985; 14(2): 142-4.
8. Dutly F, Baumer A, Kayserili H, Yüksel-Apak M, Zerova T, Hebisch G, et al. Seven cases of Beckwith-Wiedemann Syndrome, including the first reported case of mosaic paternal isodisomy along the whole chromosome 11. Am J Med Genet 1998; 79: 347-53.
9. Weng EY, Moeschler JB, Graham JM. Longitudinal observations on 15 children with Beckwith-Wiedemann Syndrome. Am J Med Genet 1995; 56: 366-73.
10. Gu D, O Dell SD, Chen XH, Müller GJ, Day IN. Evidence of multiple causal sites affecting weight in the IGF-2 INS TH region of human chromosome 11. Hum Genet 2002; 110(2): 173-81.
11. Takato T, Kamei M, Kato K, Kitano I. Cleft Palate in the Beckwith-Wiedemann Syndrome. Ann Plast Surg 1989; 22: 347.
12. Menard R, Delaire J, Schendel AS. Treatment of the Craniofacial Complications of Beckwith-Wiedemann Syndrome. Plast Reconstr Surg 1995; 96: 27.
13. Kopriva D, Classen DA. Regrowth of Tongue Following Reduction Glossoplasty in the Neonatal Period for Beckwith-Wiedemann Macroglossia. J Otolaryngol 1998; 27: 4.
1 MD, Dept. de Otorrinolaringologia, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, SP.
2 MD, Dept. de Otorrinolaringologia, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo.
3 MD, Dept. de Otorrinolaringologia, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo.
4 MD, Dept. de Otorrinolaringologia, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo.
Departamento de Otorrinolaringologia, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo -
Avenida Dr. Eneas de Carvalho Aguiar, 255, 6º andar- sala 6002 - ICHC, São Paulo.
Endereço para correspondência: Fábio Zerati - Rua Arthur de Azevedo 1445 ap. 111 Pinheiros São Paulo 05404-013.
Tel.: (0xx11) 8207-2665 - E-mail: fabio.zerati@itelefonica.com.br
Artigo recebido em 12 de fevereiro de 2004. Artigo aceito em 29 de abril de 2004.



Versão para impressão: ![]()
All rights reserved - 1933 /
2026
© - Associação Brasileira de Otorrinolaringologia e Cirurgia Cérvico Facial