INTRODUCTIONAccording to Alström et al.,
1 Alström syndrome is a genetically transmitted disease with a variety of symptoms that include progressive sensory degeneration, resulting in visual and auditory deficits, as well as obesity. Metabolic, endocrine, renal, cardiac, and hepatic dysfunctions are also observed.
Descriptions of clinical cases of Alström syndrome are scarce. The first significant scientific article was published by Eggitt Russell et al.,
2 who performed a longitudinal study of 22 cases of Alström syndrome, focusing on the histopathology of affected organs and other factors that led to the death of the affected patients. Marshall et al.
3 studied 182 cases of the syndrome. According to Welsh et al.,
4 the genetic factor that causes the syndrome was a reciprocal translocation in a novel gene of unknown function, ALMS1, located on chromosome 2p13.
Even though Alström syndrome has been described as a homogeneous alteration resulting from a mutation in a single gene, families have been described with variable clinical presentations. Cardiomyopathy, hepatic dysfunction, hypothyroidism, and hypogonadism can occur. These symptoms vary in individuals from the same family, due to the interactions of genetic modifiers.
1 Collin et al.
5,6 indicated that the phenotype of Alström syndrome is due to an altered function, rather than inadequate organ development.
Alström et al.
1 reported the onset of hearing loss in patients affected by the syndrome before the age of 10 years and noted subsequent progression. Michaud et al.
7 confirmed that deafness began in the first decade of life. Marshall et al.
3 indicated that hearing loss appeared during childhood and that the mean age of onset was 5 years. Coincidentally, chronic serous otitis media was observed in 42% of their patients, adding a conductive component to the already described sensorineural loss.
7 Russell-Eggitt et al.
2 reported that only two of their 22 cases had normal hearing after 12 years of age.
The present study aimed to perform an extensive literature review and to present a complete clinical-audiometric profile, and molecular genetic analysis of two siblings, the only two cases reported to date in South America, in order to provide a better understanding of this very rare disease.
MATERIAL AND METHODSA prospective, descriptive analytic study of the phenotype of a Caucasian Brazilian family with members affected by Alström syndrome, was conducted. Data were collected through a long-term clinical evaluation of patients, chart review, and a questionnaire answered by family members.
Patients underwent a complete otorhinolaryngological assessment with audiological examination with pure-tone and speech audiometry, acoustic immitance testing, distortion-product otoacoustic emissions (DPOAE), transient-evoked otoacoustic emission (TEOAE), and auditory brainstem response (ABR) testing.
Pure tone audiometry by air conduction was performed, at frequencies from 250 to 8,000 Hz, and by bone conduction at frequencies from 500 to 4,000 Hz; speech recognition and speech reception thresholds were measured using GSI68 equipment, equipped with TDH-39 earphones. Tympanometry was performed using GSI38 equipment. The criteria proposed by Russo and Santos were employed for this assessment.
8DPOAE and TEOAE were performed using a Biologic equipment (Scout model). Amplitudes and signal/noise ratio were evaluated in both ears, based on the criteria described by Figueiredo
9 and by Gorga.
10 ABR was performed in a Biologic equipment (Navigator-Pro model). The electrodes were placed according to the international electrode system:
11 the active electrode on the vertex (Cz); the ground, on the forehead (Fz); and the reference electrodes on the lobes of the right and left ears (A1 and A2). The headphone used in the test was the TDH-39. The type of stimulus was a click with an intensity of 80 dB HL. Absolute latencies of waves I, III, and V were measured, as well as interpeak intervals of waves I-III, III-V, and I-V, and binaural comparison of wave V latencies. The Hood criteria were used for this assessment.
12 To perform the molecular genetic diagnosis, an informed consent was obtained from all patients involved in the study and their parents/guardians. This study was approved by the research ethics committee of the institution, under protocol number 1112000005. Genomic DNA, isolated from peripheral blood according to the standard method, was amplified using the polymerase chain reaction (PCR) protocol, as previously described.
10 Sequences of primers and amplification conditions are available at request. PCR amplicons of exons 8, 10, and 16 of ALMS1 gene were purified, directly sequenced, and analyzed using an ABI 3730 Sequencing Analyzer system (Applied Biosystems - Forster City, CA, USA) system. The resulting sequences were compared with reference sequences from the GenBank mRNA (NM_015120.4). The nomenclature of the mutations was defined by den Dunnen and Antonarakis.
11RESULTSBoth affected patients had progressive sensorineural deafness first detected at approximately 6 years of age. Fig. 1 presents the family's pedigree. Computed tomography and magnetic resonance imaging (MRI) of the mastoid were performed in both patients, but revealed no abnormalities. Tests were negative for enlarged vestibular aqueduct or cochlea-saccular dysplasia.
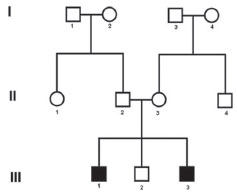
Figure 1 Pedigree showing the two cases with Alström Syndrome (case 1: III-1 and case 2: III-3).
Genetic analysis confirmed the mutation in ALMS1 gene in the two brothers. Two heterozygous mutations were identified in ALMS1 exon 10 (c.7942C > T, Gln2648* and c.9163ª > T, Lys3055*), confirming the diagnosis of Alström syndrome. (Fig. 2)
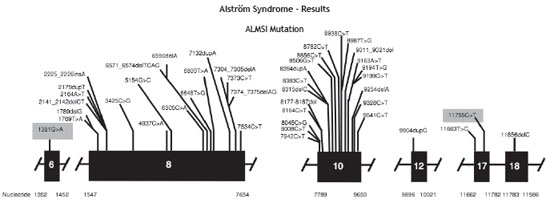
Figure 2 Mutation in the ALMS1 gene.
Patient 1 presented at age 30 years, with a history of photophobia since birth, nystagmus since age 6 months, and retinitis pigmentosa since the age of 10 months. He did not have a history of recurrent otitis media. He had dilated cardiomyopathy, diagnosed at age 28 years, but no congestive heart failure. He had no pulmonary, hepatic, or renal symptoms. He had diabetes with insulin resistance diagnosed at age 27, in addition to hyperinsulinemia, hypertriglyceridemia, hypogonadism, hypothyroidism, acanthosis nigricans, and obesity since childhood.
Patient 1 was initially diagnosed with sensorineural hearing loss of unknown genetic etiology. Cochlear function tested at age 6 years demonstrated moderate bilateral sensorineural deafness. Since then, he underwent serial pure tone and speech audiometry. Figs. 3 and 4 show the results of pure-tone audiograms at ages 21 and 30 years, respectively.
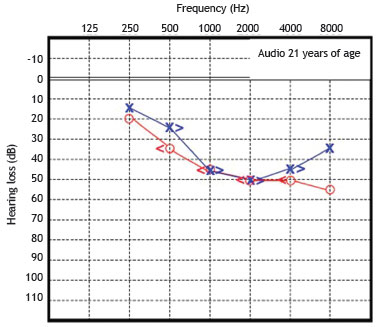
Figure 3 Audiometry in patient 1, performed at 21 years of age.
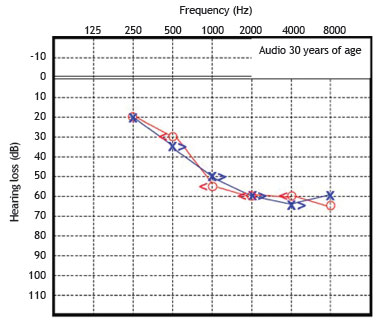
Figure 4 Audiometry in patient 1, performed at 30 years of age.
These serial audiograms document a slowly progressive, bilateral sensorineural hearing loss. At the age of 28 years, hearing impairment required the use of a hearing aid bilaterally, with partial improvement in hearing acuity in both ears.
His most recent audiometric assessment, performed at 30 years of age, showed a mean loss of 50 dB HL for the right and left ears, as shown in Fig. 3. Tympanometry showed normal "A" curves for both ears.
Recent otoacoustic emissions tests for patient 1 reveal absent DPOAE and TEOAE responses in both ears. Amplitudes and signal/noise ratio were analyzed bilaterally and were below the expected values compared with normal subjects, compatible with altered cochlear function.
ABR performed at an intensity of 80 dB HL was normal for both ears, with the presence of waves I, III, and V. The absolute latencies of waves I, III, and V, and interpeak intervals for waves I-III, III-V, and I-V were within normal limits, as was a binaural comparison of wave V latency.
Patient 2 presented at age 26 years and gave a history of, photophobia since birth, nystagmus at 6 months of life, and the onset of retinitis pigmentosa in childhood . He also had recurrent otitis with otalgia and otorrhea since early childhood,. He was diagnosed with dilated cardiomyopathy at 2 months of age, without congestive heart failure; the patient underwent treatment for one year, and the cardiomyopathy completely disappeared. He experienced no pulmonary and renal symptoms. He had liver dysfunction diagnosed at age 20 years. He did not have diabetes, but had hyperinsulinemia, hypertriglyceridemia, hypogonadism, hypothyroidism, and obesity since childhood. The patient also did not present acanthosis nigricans.
Patient 2 was diagnosed with sensorineural hearing loss of unknown genetic etiology, as was his older brother. Cochlear function at age 6 was tested, and was abnormal. Since then, this patient has undergone serial assessments, including pure-tone and speech audiometry (Figs. 5 and 6).
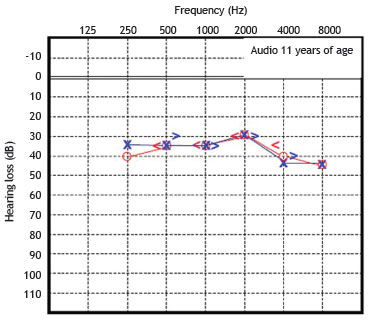
Figure 5 Audiometry in patient 2, performed at 11 years of age.
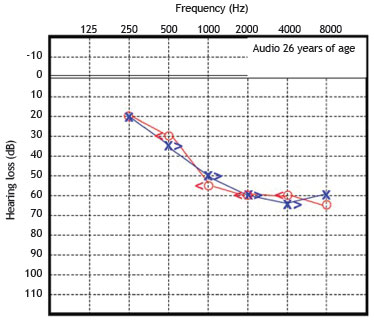
Figure 6 Audiometry in patient 2, performed at 26 years of age.
These serial audiometric assessments reveal slowly progressive, bilateral sensorineural hearing loss. At the age of 23 years, the hearing deficit required the use of bilateral hearing aids, with partial improvement in hearing acuity in both ears. The most recent audiogram, performed at age 26 years, showed a mean loss of 70 dB HL for the right ear and 65 dB HL for the left ear. Tympanometry showed the presence of "As" curves bilaterally.
DPOAE and TEOAE were absent in both ears. Amplitude and signal/noise ratios were analyzed bilaterally and were below those of normal subjects, indicating altered cochlear function.
Auditory brainstem response assessment at 80 dB HL intensity was normal in both ears, and waves I, III, and V were present. The absolute latencies of waves I, III, and V, and interpeak intervals of waves I-III, III-V, and I-V were within normal limits, as was a comparison of binaural wave V latency.
DISCUSSIONThe limited number of reports related to Alström syndrome and the few cases in the world literature reflect the rarity of this condition. The early onset of sensorineural hearing loss, which can be severe as early as the third decade of life, is usually the most striking sign of this syndrome.
This study assessed two members of a Caucasian family that had a clinical picture of slowly progressive bilateral sensorineural hearing loss that began at 6 years of age. Audiometry performed at 30 years of age in one patient and at 26 years in the other showed moderate and severe sensorineural hearing loss, respectively. DPOAE and TEOAE were absent, and the signal/noise ratio was below normal when compared with normal subjects, in both cases, while tympanometry and ABR were normal. These data suggest that the lesion causing hearing loss was cochlear.
The etiology of sensorineural hearing loss in childhood has several causes, including hereditary, infectious, and development-related causes, as well as other less common causes, such as autoimmune, traumatic, and vascular.
The hereditary causes can be divided into syndromic and non-syndromic; the latter is less prevalent.
13 Among the syndromic causes, Usher syndrome is the most important differential diagnosis of hearing loss with early onset and slow progression, associated with visual loss. What is the best evidence to suggest Alström syndrome in patients with early-onset progressive sensorineural hearing loss?
First, through serial audiometric evaluations (pure tone sensitivity) may confirm a sensorineural hearing loss that slowly progresses over 15 years to a profound hearing loss. Signs of retinopathy, easily identifiable by retinoscopy and confirmed by electroretinography, can also help in the diagnosis of the syndrome.
In addition to these audiological and visual aspects, the presence of metabolic disorders such as type 2 diabetes mellitus, hypertriglyceridemia, hypogonadism, hypothyroidism, renal and hepatic dysfunction, as well as childhood obesity , although variably manifested among affected patients, can also aid in the differential diagnosis.
Since Alström syndrome is caused by mutations in the ALMS1 gene, molecular genetic analysis must be employed in order to confirm the clinical diagnosis.
14-17 This is the first description in the Portuguese literature of two members of a family affected by Alström syndrome. While there are still no scientific publications disclosing the histopathology of the cochlea in this syndrome, the authors suggest that behind this slowly progressive degeneration of the hearing function, there is a mechanism that ultimately leads to the destruction of hair cells in the organ of Corti.
Although there is no preventive treatment for sensorineural hearing loss in patients with Alström syndrome; identification is important so family counseling can be performed. Additionally, follow-up by a multidisciplinary team can help patients achieve better educational and psychosocial development, as well as facilitate the treatment and rehabilitation of associated disorders. Early identification of hearing loss and its prompt rehabilitation through the use of bilateral hearing aid devices can provide patients with better language development and ensure that they are good candidates for hearing rehabilitation with the use of cochlear implant technology.
CONCLUSIONAfter a long-term follow-up of over 20 years, it was observed that the audiometric profile of this syndrome is characterized by early onset of sensorineural hearing loss, usually in the first decade of life with slow progression, resulting in profound hearing loss in affected individuals after ten to 20 years. The probable site of injury is cochlear, as suggested by the analysis of otoacoustic emissions and auditory brainstem responses.
Notwithstanding the rarity of the syndrome, early detection and treatment of hearing loss through the use of bilateral hearing aid devices can allow an adequate language development.
SOURCE OF FINANCIAL SUPPORTLucas Moura Viana received a doctoral grant from the National Council for Scientific and Technological Development (Conselho Nacional de Desenvolvimento Científico e Tecnológico - CNPq)
CONFLICTS OF INTERESTThe authors declare no conflicts of interest.
REFERENCES1. Alstrom CH, Hallgren B, Nilsson LB, Asander H. Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the laurence-moon-bardet-biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree. Acta Psychiatr Neurol Scand Suppl. 1959;129:1-35.
2. Russell-Eggitt IM, Clayton PT, Coffey R, Kriss A, Taylor DS, Taylor JF. Alstrom syndrome. Report of 22 cases and literature review. Ophthalmology. 1998;105:1274-80.
3. Marshall JD, Bronson RT, Collin GB, Nordstrom AD, Maffei P, Paisey RB, et al. New Alstrom syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med. 2005;165:675-83.
4. Welsh LW. Alstrom syndrome: progressive deafness and blindness. Ann Otol Rhinol Laryngol. 2007;116:281-5.
5. Collin GB, Cyr E, Bronson R, Marshall JD, Gifford EJ, Hicks W, et al. Alms1-disrupted mice recapitulate human Alstrom syndrome. Hum Mol Genet. 2005;14:2323-33.
6. Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nat Genet. 2002;31:74-8.
7. Michaud JL, Heon E, Guilbert F, Weill J, Puech B, Benson L, et al. Natural history of Alstrom syndrome in early childhood: onset with dilated cardiomyopathy. J Pediatr. 1996;128:225-9.
8. Russo ICP, Santos TMM. A Prática da Audiologia Clínica. 4º ed. São Paulo: Cortez Editora; 1993.
9. Figueiredo MS. Emissões Otoacústicas e Bera (Coleção CEFAC). São Paulo: Pulso Editora; 2003.
10. Gorga MP, Stover L, Neely ST, Montoya D. The use of cumulative distribuitions to determine critical values and levels of confiance for clinical distortion product otoacoustic emission. J Acoust Soc Am. 1996;100:968-77.
11. Klem GH, Lüders HO, Jasper HH, Elger C. The ten-twenty electrode system of the International Federation. The International Federation of Clinical Neurophysiology. Electroencephalogr Clin Neurophysiol Suppl. 1999;52:3-6.
12. Hood LJ. Clinical Applications of the auditory brainstem response. San Diego, CA: Singular Publishing Group, Inc; 1998.
13. Ruben RJ. Diseases of the inner ear and sensorineural deafness. In: Bluestone CD, Stool SE, Scheetz MD, eds. Pediatric otolaryngology, Vol. 1. Philadelphia: WB Saunders; 1990. p. 547-70.
14. Tseng CJ, Lalwani AK. Cracking the auditory genetic code: part IL Syndromic hereditary hearing impairment. Am J Otol. 2000:21:437-51.
15. Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat Genet. 2002;31:74-8.
16. Hearn T, Renforth GL, Spalluto C, Hanley NA, Piper K, Brickwood S, et al. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alström syndrome. Nat Genet. 2002;31:79-83.
17. Marshall JD, Hinman EG, Collin GB, Beck S, Cerqueira R, Maffei P, et al. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alström syndrome. Hum Mutat. 2007;28:1114-23.
1. Faculdade de Medicina, Universidade de Brasília (UnB), Brasília, DF, Brazil
2. Post-graduate Program, Faculdade de Ciências da Saúde, Universidade de Brasília (UnB), Brasília, DF, Brazil
3. Instituto Brasiliense de Otorrinolaringologia, Brasília, DF, Brazil
4. The Jackson Laboratory, Maine, USA
F.B. Júnior
E-mail:
fayez@unb.brReceived 4 November 2012.
Accepted 30 August 2013.
* Study conducted at Faculdade de Ciências da Saúde, Universidade de Brasília, Brasília, DF, Brazil.

