INTRODUCTIONIt is well known that in developed countries one in 750 births is likely to produce a child with sensorineural hearing impairment and it is estimated that one in 1000 children is affected by severe deafness at birth or by the end of the prelingual period1. Approximately 60% of all causes of prelingual deafness can be attributed to genetic factors. Therefore, the genetic etiology is becoming increasingly relevant in cases of hearing impairment and/or deafness. The remaining 40% are distributed over a vast variety of etiologies2. There are three identifiable kinds of inheritance pattern for the inherited deafness: autosomal recessive, autosomal dominant, and X-linked. From 75% to 85% of cases of non-syndromic prelingual deafness are manifested as autosomal recessive forms. Autosomal dominant forms account for about 15% to 25% of cases, and the remaining 1% to 3% are of X-linked Mendelian inheritance. There are also descriptions of forms which are inherited exclusively from the mother, corresponding to mitochondrial inheritance, associated or not with autosomal dominant inheritance2,3.
In terms of phenotype, the autosomal recessive forms are more severe, being responsible for almost all forms of congenital deafness. These, in turn, are caused in great part by cochlear defects, leading to sensorineural hearing impairment. The autosomal dominant forms seem to contribute more to cases of postlingual deafness, which are usually progressive, and the impairment is, in most cases, conductive or mixed (conductive and sensorineural)4,5.
Mutations in the conexin 26 or GJB2 - Gap Junction Protein Beta 2 gene, located on the long arm of chromosome 13 (13q11-12), appear to be extremely common in the genesis of inherited non-syndromic deafness, and account for 34% to 50% of autosomal recessive sensorineural deafness (DFNB1) and for 10% to 37% of sporadic cases6-10.
Deletion of a guanine out of a sequence of six guanines going from position 30 to position 35 in the conexin 26 (GJB2) gene is the mutation that occurs in 60% to 80% of cases. This nucleotide deletion can occur at position 35 (35delG) or at position 30 (30delG) of the gene, but the deletion at position 35 (35delG), genetically related to chromosome 13, is the more frequent one, varying between approximately 50% to 85% of the non-syndromic deafness cases in patients from Italy, Spain, and Israel7,11-13. In a study with Brazilian patients14, mutations in gene GJB2 were found in 22% of families tested with at least one patient with hearing loss, and in 11.5% of cases in which an environmental etiology was not completely ruled out. In the cases in which this mutation in conexin 26 gene occurs, the gene product, a protein named conexin, no longer exerts its functions correctly, while the cochlea is structurally normal15.
Even though the GJB2 gene is of great importance in the etiology of non-syndromic sensorineural impairment, there are so far very few studies describing the audiometric characteristics of patients with mutations in the GJB2 gene associated with hearing loss, especially in Brazil. According to these studies, in patients who are homozygous for mutations in the GJB2 gene and mainly in those who are homozygous for 35delG, the hearing loss is characterized for being prelingual, affecting all frequencies, being non-progressive, varying in degree from moderate to deep, even among impaired siblings of the same family, and not being associated with vestibular alterations and radiological abnormalities of the inner ear16,17. The present study had the objective of analyzing the audiometric characteristics of patients with mutations in the conexin 26 gene, in order to outline a genotype-phenotype correlation.
PATIENTS AND METHODDuring the period from March through June, 2000, a cross-section study was carried out, in which 33 index cases (23 male and 10 female) with non-syndromic sensorineural hearing impairment were investigated. They aged between 3 and 37 years, were randomly selected at the Ear-Nose-Throat Outpatient Clinic. Of these index cases, eight relatives were evaluated (4 male and 4 female), aged 11 to 45 years, who also presented non-syndromic sensorineural hearing impairment. Hence, we evaluated 33 families with at least one member with hearing loss, totaling 41 impaired individuals. This study was approved by the Research Ethics Committee, Protocol No. 4429/2000. The complete history of each patient was obtained to investigate age at onset of the hearing impairment, presence of other cases in the family, and to exclude the possibility of environmental causes such as, maternal-fetal infections, perinatal complications, meningitis, use of ototoxic drugs, acoustic trauma. Physical, otorhinolaryngological and systemic examinations, as well as complementary tests, were carried out to exclude signs suggesting syndromic forms of hearing loss (especially craniofacial dysmorphism, skin disorders, anomalies of branchial, cardiac or thyroidal origin, vision disorders, etc.). Moreover, patients were submitted to ophthalmologic evaluation (including fundoscopy), vestibular tests and computerized tomography of the temporal bone. Thus, a complete clinical evaluation was performed to exclude patients with hearing loss caused by environmental factors, congenital malformations of the inner ear or genetic syndromes. The patients were audiologically tested by pure-tone audiometry, performed at the Speech Therapy Outpatient Clinic, and those with non-syndromic sensorineural hearing impairment classified as mild (25-40 dB), moderate (41-60 dB), severe (61-80 dB) or profound (>81 dB) were included18.
Molecular analysis was carried out at the Molecular Biology Center, after DNA extraction from whole blood, done at the Genetics and Molecular Biology Research Unit, using a genomic DNA extraction kit (GFXTM Genomic Blood DNA Purification Kit, Amersham Pharmacia Biotech Inc.), according to the manufacturer protocol. To detect the 35delG mutation, the technique used was allele-specific polymerase chain reaction (AS-PCR) with specific primers8. Primers named control A (direct) and B (reverse) were also synthesized, for co-amplification of the GJB2 gene with a segment of the amelogenin gene homologous to the X-Y chromosome, thus being used as internal amplification controls19. Additionally, the PCR technique was used to detect the Delta (GJB6 - D13S1830) mutation, with specific primers20, in heterozygous samples and in those who did not present the 35delG mutation. The AS-PCR and PCR products were analyzed by electrophoresis on 1.5% agarose gel in TBE 1X buffer, containing ethidium bromide, at a concentration of 0.5mg/mL, submitted to ultraviolet light to confirm that the reaction was successful, and the gel was photographed. The samples which did not present the mutations under study on both alleles, or heterozygous samples, were submitted to automated sequencing.
Two pairs of primers were synthesized21 and amplification of the GJB2 gene in two fragments was obtained by the PCR reaction prior to sequencing. The amplified fragments were purified using the Wizard SV Gel and PCR Clean-UP System Kit (PROMEGA), and the sequencing reactions were run in both directions in an ABI PRISMTM 377 automated sequencer (Perkin Elmer) using the BigDyeTM Terminator Cycle Sequencing Kit V2.0 Ready Reaction (ABI PRISM/PE Biosystems). The sequences obtained were analyzed and compared with the normal sequence, using the Gene Runner V3.05 program to align the nucleotide sequences and the Chromas V1.45 program to edit the electropherograms.
The present retrospective study was submitted to and approved by the Research Ethics Committee, Protocol No. 2813/2004, in order to review the molecular and audiometric data of patients.
Percentages with their respective standard deviations were calculated, and the results are expressed in % (SD%).
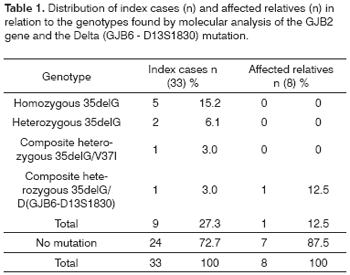
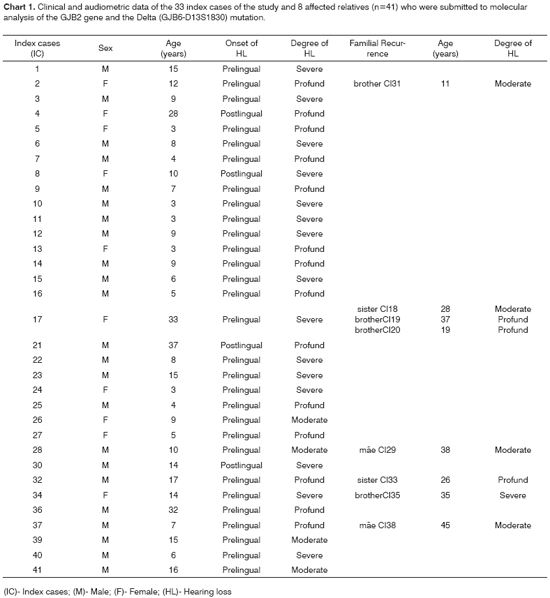
RESULTSTable 1 presents the overall genotype results obtained after molecular analysis performed in the 33 index cases and 8 affected relatives. The clinical and audiometric data concerning the index cases, such as gender, age, time of onset and degree of hearing loss, and familial recurrence, are shown in Chart 1.
Therefore, we found a prevalence of 27.3% (SD%=7.7) for the 35delG mutation in the index cases analyzed (9/33), of 21.2% (SD%=5.03) of the alleles (14/66) with the 35delG mutation, and of 12.5% (SD%=11.69) in the affected relatives. For each mutation, V37I and Delta (GJB6-D13S1830), a 3% prevalence (SD%=2.96) was found.
The audiometric tests performed in patients with hearing impairment (n=41) showed the following results with regard to degree of loss: profound - 17 patients (41.5%, SD%=7.69); severe - 16 patients (39.0%, SD%=7.61) and moderate - 8 patients (19.5%, SD%=6.18). Of the five patients who were homozygous for 35delG, one presented a profound degree (2.43%, SD%=2.40), three presented a severe degree (7.31%, SD%=4.06), and one patient presented a moderate degree of hearing loss (2.43%, SD%=2.40). Of the two patients who were heterozygous for 35delG, one (2.43%, SD%=2.40) presented a severe and the other (2.43%, SD%=2.40) a moderate degree of hearing loss. The patient who was a 35delG/V37I composite heterozygote (2.43%, SD%=2.40) and the two 35delG/Delta (GJB6-D13S1830) composite heterozygotes (4.87%, SD%=3.36) presented a severe degree of hearing loss. The predominant audiometric frequencies were 4000 to 8000 Hertz (Hz). All patients with the 35delG mutation presented hearing loss with prelingual onset.
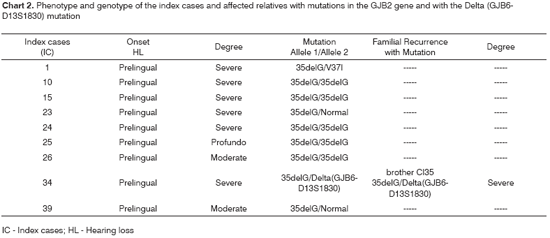
The phenotype/genotype correlation of the index cases and affected relatives, with the diagnosed mutation, is represented in Chart 2.
DISCUSSIONIn the present study, we found a 27.3% prevalence of the 35delG mutation in the index cases analyzed, 21.2% of alleles with the mutation, and 12.5% in the affected relatives. A prevalence of 3% was also assessed for each one of the mutations V37I and Delta (GJB6-D13S1830), found in the study. These results are concordant with previous studies reported in the literature, conducted in several populations22-26. The relative contribution of the 35delG mutation to the non-syndromic hearing loss in these populations varied from 0% (Oman, Korea, Japan) to 70% (Italy, Spain, Greece), demonstrating the genetic heterogeneity among the different countries, even though some of these studies were based on a small number of patients, besides the differences in the impairment investigation criteria and the mutation screening methods23,27-30.
Recent studies found a 342 thousand base-pair (342 Kb) deletion close to the GJB6 [D(GJB6 - D13S1830)] gene, suggesting that this mutation could cause non-syndromic recessive hearing loss, either by a homozygous deletion or by digenic penetrance of the deletion in the GJB6 gene, associated with a trans mutation in the GJB2 gene in the heterozygous cases24. Most genetic cases of hearing impairment result from mutations in a single gene, but an increasing number of cases with two involved genes have been identified31. A multicenter study conducted in nine countries demonstrated that the Delta (GJB6-D13S1830) mutation is more frequent in France, Spain, Israel, the United Kingdom and Brazil, varying from 5.9% to 9.7% of all studied alleles of patients with DFNB1, and being present in about 50% of heterozygous patients in Spain32. In the present study, a 3% prevalence of the deletion in the GJB6 gene in heterozygosis with the 35delG mutation was found, which is in agreement with data from the literature24,32.
In Brazil, a study conducted in newborns from the region of Sao Jose do Rio Preto, SP33, found a 2.24% (1:44.6) prevalence of heterozygotes for the 35delG mutation, and another newborn screening, performed in the region of Campinas, SP34, found a 0.97% (1:103) prevalence of heterozygotes. In another study, performed in patients with hearing loss, mutations in the GJB2 gene were found in 33.5% of cases, and only the 35delG mutation was identified in 84.2% of mutant alleles14. The methodology used in the current study, AS-PCR and automated sequencing, was similar to the three above mentioned researches, but a variation was found in the frequency of the alleles with the 35delG mutation. This finding can be explained by differences in the sample or maybe by the highly heterogeneous ethnic composition of the Brazilian population, with miscegenation among various ethnic groups, mainly Caucasoid and African, allowing the occurrence of differences in prevalence among different regions of the country35.
According to the literature, analyses of the GJB2 gene in patients with hearing impairment frequently demonstrate heterozygosis in about 10% to 42% of cases, in spite of the fact that most of the mutations are recessive23,36,37. In this study, we found a 12.1% frequency of heterozygous index cases, a result that agrees with the literature23,36,37.
According to the audiometric results, in the present study the hearing loss was profound in 41.5% of patients, severe in 39.0%, and moderate in 19.5%, with a predominance of the high frequencies (4000-8000 Hz). In patients who were homozygous or heterozygous for 35delG, the moderate-severe degrees of hearing loss were predominant, a pattern that is in agreement with the literature17,38,39. Patients who are homozygous for 35delG display a large variability in the degrees of hearing loss. Most of the autosomal recessive impairments are phenotypically rather consistent, even among sibs, a feature that is not observed for the GJB2-35del gene, mainly in heterozygous cases. This suggests the possibility of other factors modulating the expression of the mutant gene40. An intriguing possibility is that there may be a second conexin gene sharing functions with the GJB2 gene. It is conceivable that a second conexin protein might act as a substitute under certain conditions. Maybe there are modifier genes at other locations or environmental influences which activate or inactivate the promoter regions of the gene. As protein Cx26 is involved in the ion homeostasis of the inner ear, some of these patients may be capable of hearing, for they present a moderate hearing loss, suggesting the existence of alternative or compensatory homeostatic mechanisms. Alterations in protein Cx26 can adversely affect the development of the hearing system, resulting in variations of the degree of hearing loss or assymmetry38. Environmental influences, such as noises and ototoxic drugs, can be additive or synergists to the defects caused by mutations in the GJB2 gene, thus increasing the hearing loss17,38,41.
The clinical evaluations of patients in this study do not suggest that environmental causes are the main factors, considering that an autosomal recessive transmission pattern was identified in cases with the 35delG mutation. Moreover, upon audiometric testing, these nine patients presented non-progressive audiometric findings, similar to those of patients described in the literature23,42-44 diagnosed with hearing impairment caused by mutation in the GJB2 gene. This confirms that the gene is involved in the etiology of the hearing loss of patients of this study with a phenotypic expression similar to that described in the literature23,42-44. In approximately one third of the cases of hearing impairment due to mutations in the GJB2 gene, an audiometric pattern of progressive hearing loss is found, as opposed to the two thirds of cases with the typical non-progressive pattern. This means that a child with moderate hearing loss may progress to profound, and the therapies for these two kinds of degrees of hearing loss may be different. Families with a child with moderate-severe or profound hearing loss can be benefited by an analysis of the GJB238 gene.
The 35delG mutation is easy to detect and the test is viable. However, since a great part of the patients with the 35delG mutation are homozygous, broader analyses of the GJB2 gene will be necessary in a high proportion of cases, in order to distinguish the common heterozygous carriers (healthy carriers) from the heterozygous patients with DFNB1. These broader analyses and investigations [including analyses of the entire coding region, the promoter region, the non-coding region of the gene and analyses of the Delta (GJB6-D13S1830) mutation], as performed in the present study, should be oriented by the clinical characteristics of DFNB143,45.
According to the literature, molecular tests associated to audiometric data can predict that a significant number of patients with the 35delG mutation will present a moderate-severe hearing loss and others are expected to present profound hearing loss29,44,46, as found in this study. Thus, in spite of the small casuistic, the audiometric pattern was concordant with the literature, enabling us to establish a genotype-phenotype correlation in the ten patients of the sample (9 index cases and 1 affected relative), i.e., the patients with the 35delG mutation presented a moderate-severe to profound, non-progressive hearing loss. A multicenter study is however necessary to verify the true phenotypic expression related to the 35delG mutation in the Brazilian population. Knowledge of the genotype will enable physicians, speech therapists, educators, helped by clinical geneticists, to provide more adequate counseling to parents by evaluating the risk of a future child having a similar hearing impairment. Children diagnosed before six months of age and submitted to a successful treatment with amplification will have a much greater chance of normally developing speech and language.
REFERENCES 1. Morton NE. Genetic epidemiology of hearing impairment. Ann N Y Acad Sci 1991;630:16-31.
2. Mustafa T, Arnos KS, Pandya A. Advances in hereditary deafness. Lancet 2001;358:1082-90.
3. Skvorak Giersch AB, Morton CC. Genetic causes of nonsyndromic hearing loss. Curr Opin Pediatr 1999;11(6):551-7.
4. Petit C. Genes responsible for human hereditary deafness: symphony of a thousand. Nature Genet 1996;14:385-91.
5. Van Camp G, Willems PJ, Smith RJH. Nonsyndromic hearing impairment: unparalleled heterogeneity. Am J Hum Genet 1997;60:758-64.
6. Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang, JN, Parry G, Mueller, RF, Leigh IM. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997;387:80-3.
7. Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, Kimberling WJ. Novel mutations in the connexin 26 gene (GJB2) that cause autossomal recessive (DFNB1) hearing loss. Am J Hum Genet 1998;62:792-9.
8. Scott DA, Kraft ML, Carmi R, Ramesh A, Elbedour K, Yari Y, Srisailapathy CRS, et al. Identification of mutation on the connexin 26 gene that cause autossomal recessive nonsyndromic hearing loss. Hum Mutat 1998;11:387-94.
9. Gabriel H, Kupsch P, Sudendey Jr, Winterhager E, Jahnke K, et al. Mutations in the connexin 26/GJB2 gene are the most common event in non-syndromic hearing loss among the German population. Hum Mutat 2001;17:521-2.
10. Van Camp G, Smith RJH. Na Hereditary Hearing Loss Homepage [Site na Internet]. Disponível em: http://webhost.ua.ac.be/hhh/. Acessado em 2006.
11. Zelante L, Gasparini P, Estivill X, Melchionda S, D'Agruma L, Govea N, Mila M, Della Monica M, et al. Connexin 26 mutations associated with the most common form of non-syndromic neurosensory autossomal recessive deafness (DFNB1) in Mediterraneans. Hum Molec Genet 1997;6:1605-9.
12. Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, D'Agruma L, Mansfield E, Rappaport E, et al. Connexin 26 mutations in sporadic and inherited sensorineural deafness. Lancet 1998;351:394-8.
13. Antoniadi T, Gronskov K, Sand A, Pampanos A, Brondum-Nielsen K, Petersen MB. Mutation analysis of the GJB2 (connexin 26) gene by DGGE in greek patients with sensorineural deafness. Hum Mutat 2000;16:7-12.
14. Oliveira CA, Maciel-Guerra AT, Sartorato EL. Deafness resulting from mutations in the GJB2 (connexin 26) gene on Brazilian patients. Clin Genet 2002;61:354-8.
15. Kammen-Jolly K, Ichiki H, Scholtz AW, Gsenger M, Kreczy A, Schrott-Fischer A. Connexin 26 in human fetal development of the inner ear. Hear Res 2001;160(1-2):15-21.
16. Denoyelle F, Marlin S, Weil D, Moatti L, Chauvin P, Garabedian EN, Petit C. Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin 26 gene defect: implications for genetic counselling. Lancet 1999;17(9161):1298-303.
17. Cryns K, Orzan E, Murgia A, Huygen PLM, Moreno F, del Castilo I, et al. A genotype-phenotype correlation for GJB2 (connexin 26) deafness). J Med Genet 2004;41:147-54.
18. World Health Organization. Report of the informal working group on prevention of deafness and hearing impairment programme planning. Geneva: WHO, 1991. 22p
19. Antoniadi T, Gronskov K, Sand A, Pampanos A, Brondum-Nielsen K, Petersen MB. Mutation analysis of the GJB2 (connexin 26) gene by DGGE in Greek patients with sensorineural deafness. Hum Mutat 2000;16:7-12.
20. del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, et al. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 2002;346:243-9.
21. Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, Kimberling WJ. Novel mutations in the connexin 26 gene (GJB2) that cause autossomal recessive (DFNB1) hearing loss. Am J Hum Genet 1998;62:792-9.
22. Sobe T, Vreugde S, Shahin H, Berlin M, Davis N, et al. The prevalence and expression of inherited connexin 26 mutations associated with non-syndromic hearing loss in the Israeli population. Hum Genet 2000;106:50-7.
23. Wilcox SA, Saunders K, Osborn AH, Arnold A, Wunderlich J, et al. High frequency hearing loss correlated with mutations in the GJB2 gene. Hum Genet 2000;106:399-405.
24. del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, et al. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 2002;346:243-9.
25. Frei K, Szuhai K, Lucas T, Weipoltshammer K, Schofer C, Ramsebner R, et al. Connexin 26 mutations in cases of sensorineural deafness in eastern Austria. Eur J Hum Genet 2002;10:427-32.
26. Pampanos A, Economides J, Iliadou V, Neou P, Leotsakos P, Voyiatzis, et al. Prevalence of GJB2 mutations in prelingual deafness in the Greek population. Int J Pediatr Otorhinolaryngol 2002;65:101-8.
27. Gasparini P, Estivill X, Volpini V, Totaro A, Castellvi-Bel S, et al. Linkage of DFNB1 to non-syndromic neurosensory autosomal-recessive deafness in Mediterranean families. Eur J Hum Genet 1997;5:83-8.
28. Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, D'Agruma L, Mansfield E, Rappaport E, et al. Connexin 26 mutations in sporadic and inherited sensorineural deafness. Lancet 1998;351:394-8.
29. Kenna MA, Wu B-L, Cotanche DA, Korf BR, Rehm HL. Connexin 26 studies in patientes with sensorineural hearing loss. Arch Otolaryngol Head Neck Surg 2001;127:1037-42.
30. Simsek M, Al-Wardy N, Al-Khayat A, Shanmugakonar M, Al-Bulushi T, Al-Khabory M, et al. Absence of deafness associated connexin 26 (GJB2) gene mutations in the Omani population. Hum Mutat 2001;18:545-6.
31. Nance WE. The genetics of deafness. Ment Retard Disabil Res Rev 2003;9:109-19.
32. del Castillo I, Moreno-Pelayo MA, del Castillo FJ, Brownstein Z, Marlin S, Adina Q, et al. Prevalence and Evolutionary Origins of the del(GJB6-D13S1830) Mutation in the DFNB1 Locus in Hearing Impaired Subjects: a Multicenter Study. Am J Hum Genet 2003;73:1452-8.
33. Piatto VB, Oliveira CA, Alexandrino F, Pimpinati CJ, Sartorato EL. Perspectivas para triagem auditiva genética: rastreamento da mutação 35delG em neonatos. J Pediatr 2005;81:139-42.
34. Sartorato EL, Gottardi E, Oliveira CA, Magna LA, Annichio-Bizzacchi JM, Seixas CA, Maciel-Guerra AT. Determination of the frequency of the 35delG in Brazilian neonates. Clin Genet 2000;58:339-40.
35. Oliveira CA, Alexandrino F, Abe-Sandes K, Silva Jr WA, Maciel-Guerra AT, Magna LA, Sartorato EL. Frequency of 35delG in the GJB2 gene in samples of Caucasians, Asians and African Brazilians. Hum Biol 2004;76:313-6.
36. Pandya A, Arnos KS, Xia XJ, Welch KO, Blanton SH, Friedman TB, et al. Frequency and distribution of GJB2 (connexin 26) and GJB6 (connexin 30) mutations in a large North American repository of deaf probands. Genet Med 2003;5:295-303.
37. Stevenson VA, Ito M, Milunsky JM. Connexin-30 deletion analysis in connexin-26 heterozygotes. Genet Test 2003;7:151-4.
38. Cohn ES, Kelley PM, Fowler TW, Gorga MP, Lefkowitz, et al. Clinical studies of families with hearing loss attributable to mutations in the connexin 26 gene (GJB2/DFNB1). Pediatrics 1999;103:546-50.
39. Murgia A, Orzan E, Polli R, Martella M, Vinazi C, Leonardi E, Arslan E, Zacchello F. Cx26 deafness: mutation analysis and clinical variability. J Med Genet 1999;36:829-32.
40. Marlin S, Garabedian E-N, Roger G, Moatti L, Matha N, Lewin P, Petit C, Denoyelle F. Connexin 26 gene mutations in congenitally deaf children. Arch Otolaryngol Head Neck Surg 2001;127:927-33.
41. Rabionet R, Zelante L, Lopez-Bigas N, DAgruma L, Melchionda S, Restagno G, et al. Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum Genet 2000;106:40-4.
42. Cohn ES, Kelley PM. Clinical phenotype and mutations in connexin 26 (DFNB1/GJB2), the most commom cause of childhood hearing loss. Am J Med Genet 1999;89:130-6.
43. Denoyelle F, Marlin S, Weil D, Moatti L, Chauvin P, Garabedian EN, Petit C. Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin 26 gene defect: implications for genetic counselling. Lancet 1999;17:1298-303.
44. Engel-Yeger B, Zaaroura S, Zlotogora J, Shalev S, Hujeirat Y, Carrasquilo M, Barges S, Pratt H. The effects of a connexin 26 mutation - 35delG - an oto-acoustic emissions and brainstem evoked potentials: homozygotes and carriers. Hear Res 2002;163:93-100.
45. Mustapha M, Salem N, Delague V, Chouery E, Ghassibeh M, Rai M, Loiselet J, Petit C, Megarbane A. Autosomal recessive non-syndromic hearing loss in the Libanese population: prevalence of the 30delG mutation and report of two novel mutations in the connexin 26 (GJB2) gene. J Med Genet 2001;38:e36.
46. Yoshinaga-Itano C, Sedey AL, Coulter DK, Mehl AL. Language of early-and later-identified children with hearing loss. Pediatrics 1998;102:1161-71.
1 PhD, Adjunct Professor - Department of Otorhinolaryngology / Head and Neck Surgery - FAMERP.
2 5th Year Medical Student - FAMERP.
3 M.Sc. Head of the Speech and Hearing Therapy Division - Department of Otorhinolaryngology / Head and Neck Surgery - FAMERP.
4 Associate Professor - Head of the Department of Otorhinolaryngology / Head and Neck Surgery - FAMERP.
5 M.Sc. Department of Otorhinolaryngology / Head and Neck Surgery - FAMERP.
6 PhD. Head of the Center for Molecular Biology and Genetic Engineering - CBMEG-UNICAMP.
Faculdade de Medicina de São José do Rio Preto, SP - FAMERP.
Send correspondence to: Vânia Belintani Piatto - Rua Santina Figliagi Ceccato 450 ap 23-A. Vila Itália São José do Rio Preto SP 15.035-180.
BIC-FAMERP (Bolsa de Iniciação Científica - FAMERP).
Paper submitted to the ABORL-CCF SGP (Management Publications System) on August 31th, 2006 and accepted for publication on November 2nd, 2006. cod. 3367.