INTRODUCTIONOral squamous cell carcinoma (OSCC) also named epidermoid carcinoma, amounts to over 90% of all malignant tumors that affect the oral cavity 1. It affects mainly male patients aged 50 to 80 years 1. The smoking and alcohol abuse are well-established risk factors in most cases 1. However, a small proportion of cases (15-20%) occurs in patients without history of smoking or alcohol abuse, suggesting the presence of other risk factors 2.
An important fact learned from epidemiological studies is that there has been an increase in development of OSCC in patients younger than 45 years of age as of the 80's 3, 4. The behavior of the tumor in young adults is controversial, and there are reports of greater anaplasia and worse prognosis, comparing to the habitual age range 1, 3-5. Additionally, the implication of the agents with known carcinogenic potential such as tobacco and alcohol in this group is poorly defined and normally not reported 1, 3-5. Therefore, other factors have been investigated, especially of microbial and molecular nature 1, 3-5.
A comparison of OSCC cases in patients younger than 40 years of age between the 60 and 70's and our current year shows that the incidence of OSCC in young adults has doubled, reaching 6% of all cases of OSCC 4. This fact coincides with more concern about epidemiological surveys and case reports. However, comparisons between the 80's and 90's demonstrate the same increase in rate 3.
In the US, mortality rate of OSCC has decreased in Caucasian older men, possibly related with reduction of smoking in the population 3. Conversely, mortality rate by tongue OSCC in young male patients has increased, demonstrating poor response to treatment 3.
One of the main causes that leads to limitation of response to treatment, poor prognosis and reduction of survival rates in patients with cancer is late diagnosis 10. In cases of OSCC, the delay is reflected in malpractice lawsuits filed against healthcare professionals in the USA 10. The main complaints reported are inappropriate management of patients, negligence in diagnosing, failure to perform diagnostic procedures (biopsy), delay in referring the patient to the appropriate professional, and surgical complications 10. The most common complaint - negligence in diagnosing - amounts to over 86% of the lawsuits in which the plaintiff prevails, if delay is equal or greater than 3 months 10. The term "delay in diagnosis" is not clearly defined being that in most recent studies and lawsuits, it is accepted as a minimum period of 3 months 10. However, this time tends to be longer in younger patients, since less experienced professionals do not consider the hypothesis of malignancy and tend to treat the lesions as if they were benign 10.
According to the study by Lydiatt (2002), 45% of lawsuits of patients with OSCC are filed against dental surgeons 10. Among them, 60% reported that they should have had but have not conducted biopsy 10. According to the author, it happens because dental surgeons are not comfortable to conduct this procedure. Patients also stated that Otorhinolaryngologists failed to make early cancer diagnosis, but they conducted biopsy and more frequently referred the patients to specialized treatment 10. However, owing to the fact that dental practice is less related with malignant conditions, dentists manage to get a defense line in most of the lawsuits when compared to Otorhinolaryngologists 10.
In addition, it is important to refer that mean patient age that comes to legal support is 45 years, that is, young to OSCC 10. Despite the implications of smoking and alcohol abuse as important risk factors for OSCC, they are reported only by a small percentage of young patients, as previously said, and even in cases in which there is correlation, it is said that in this group exposure to carcinogenic factors would be short and not long enough for a malignant lesion to develop 3-5, 10, 11. Moreover, a large number of people are exposed to such risk factors, such as immune and nutritional deficits, genetic factors and participation of microbiological agents. Among them, the contribution of viruses with known oncogenic potential, such as human papillomavirus (HPV) and Epstein-Barr virus (EBV) has been considered, which have been associated with OSCC for the past 20 years 8, 13.
Taking into account the need to have further clarification about biological characteristics and, in special, etiological characteristics of OSCC in adults and young adults, we proposed an analysis of information referring to etiological factors normally associated with mouth cancer such as human papillomavirus (HPV), Epstein-Barr virus (EBV) and protein p53. We also studied the participation of telomerase, one of the enzymes that support maintenance of cell replication capability 6-8. The increase in telomerase expression has been described in OSCC and in malignant tumors of other sites such as uterus, lung, liver, breast, and others 6, 9.
LITERATURE REVIEWViral Role
The cancer multifactorial etiology is widely accepted, however, in addition to the role of environmental and/or genetic factors, infectious etiology has been advocated 14. Initially, syphilis was implicated in the etiology of OSCC and, subsequently, Candida species were involved; however, recently the interest has been directed to viral etiology of OSCC 1.
The balance between proliferation and cell death (apoptosis) is vital for survival of any live organism, and this balance is ruptured by viral infection, which can lead to neoplastic transformation 14. Many genetic products of viral origin can bind to proliferation regulating genes and cell death and affect their functions 14. The initial sign of apoptosis can be inhibited by adenovirus proteins (E1B 19 kDa and E3), mixomavirus (MT2), bacillovirus (iap), simple herpes virus and cytomegalovirus, and induced by mixovirus and hepatitis C virus 14. In addition, sign transduction to cell death (p53, pRb, bcl-2) and transactivation of protooncogenes (c-myc, c-fos, c-jun) can be inhibited by adenovirus proteins, EBV, cytomegalovirus, among others 14. As a consequence, host cells with latent viral infection are lead to proliferation without regulation by cell repair or even mechanisms to eliminate damaged cells 14. These cells can continue to accumulate mutations induced by other mechanisms capable of causing DNA damage, such as tobacco, carcinogenic factors, toxins and others, which can result in development of malignant neoplasm 14.
Human Papillomavirus (HPV)
HPV is a small, non-enveloped DNA virus of family papovaviridae, with tropism for epithelial cells that can induce hyperplasia, papillomatosis and verrucuous lesions in the squamous stratified epithelium of skin and mucosa 8, 15. Over 100 different types of HPV have been identified in humans 8, 15, 16. Some of them, such as HPV 16, 18, 33 and 58, seem to have an important role in the development of some human tumors, being considered of high risk 8, 12, 15, 17.
HPV infection represents an extremely significant risk factor for cervical cancer 8, 12, 13, 15-18. High risk HPV are the main cause of anogenital cancers and have been implied in head and neck cancer cases, since the presence of viral DNA has been identified in these tumors 8, 12, 15, 17. However, its role in OSCC has not been properly defined yet 12, 15-18.
HPV infection is normally identified by viral DNA detection in cells and tissues; however, as HPV causes focal infection there are a number of frequent mistakes such as false positives and false negatives, especially in asymptomatic subjects 17. However, since HPV infection is transient, absence of HPV DNA does not rule out previous infection 17. Antibodies for antigens of HPV capsidium demonstrated they are reliable markers of previous and/or present infection and they are used in serum studies, in which detection is normally more reliable 8, 17.
Since 1977, over 600 cases of oral carcinoma have been studied by identification of HPV 18. The referred prevalence mentioned by different techniques varies from 6% to 94%, being that the most sensitive method to detect HPV seems to be polymerase chain reaction (PCR) 8, 15, 16, 19. Despite that, the mean prevalence is smaller than in cervical cancer 12, 18. However, similarly to the genital tract, HPV-16 and 18 are by far the most frequently found, amounting to 80% of positive HPV lesions 12, 17, 18.
High risk HPV is capable of immortalizing oral and cervical epithelial cells in vitro 17. Oncoproteins E6 and E7 are the two main viral oncogenes expressed in the neoplastic tissue and both stimulate cell proliferation, being capable of binding and inactivating tumor suppressant cells of the host, such as p53 and pRb, respectively 20. E6 can also activate telomerase, another factors involved in carcinogenesis 8, 16. Thus, viral oncoproteins are capable of transforming human primary keratinocytes of genital and respiratory tract causing imbalance of cell cycle regulating mechanisms, leading to genetic progression to OSCC 8, 17. Consequently, HPV infection can represent an alternative, functionally comparable to molecular mechanisms of carcinogenesis 2.
An increasing trend of positive responses to HPV in non-alcohol abusers and non-smokers has been observed when compared to users of such substances 2. Most of the studies, though, agree that there is no statistical correlation between prevalence of HPV and history of smoking, even though both participate in oral mucosa carcinogenesis 15.
An unexpected fact that has been reported is the reduction of approximately 40% of the risk of death in patients with HPV positive tumors 2. These tumors are less associated with alcohol and smoking and the principle of field cancerization does not apply since HPV infections tend to be focal, which primarily should lead to poor prognosis, which is not reported 2. Positive HPV carcinoma seems to be a distinct entity (affect more basal cells and have less inflammatory components), with distinct biology (less mutation of p53), different risk factors (less association with tobacco and alcohol) and different clinical course (greater survival) 2. The greater prevalence of positive OSCC is for HPV type 16, which is twice higher than in control groups, and there is no difference in positivity between men and women 2. There is also greater positivity for HPV in young adults and this fact would be responsible for a biological difference, in which case points to better prognosis, a fact contrary to what would be expected 16.
A study demonstrated correlation between detection by PCR of serum and lesional HPV and it was observed that patients that presented lesional HPV presented serum HPV only in more advanced stages of the disease, being that four out of six patients presented distant metastases and 50% of the patients with serum HPV died 19. However, 86% of the patients that presented lesional HPV and absence of serum HPV were free from disease evidence 19. Thus, presence of serum HPV was correlated with more advanced stages of the disease and worse prognosis 19.
There seems to be relation between local infection by HPV and detection in other sites 18. HPV has the potential to invade other epithelial cells after primary infection of epithelial site 18. Premoli-De-Percoco et al. (1998), using in situ hybridization in women with OSCC demonstrated simultaneous presence of HPV in the cervicovaginal cytology and in OSCC in 23 out of 28 cases 18. In this case, positive response of HPV in OSCC was greater in the age range 50-69 years, a group in which we would expect less prevalence of HPV 18. The natural history of the infection by HPV demonstrates a peak between sexually active women (15-25 years) and it tends to stabilize after the age of 30 years, owing to immune response, when women generally experience resolution of infection. However, others suffer small epithelial dysplasia and after a long period of incubation, combined with environmental factors, it peaks to malignant transformation of epithelial cells 18.
Epstein-Barr virus (EBV)
EBV, member of the group of human herpes virus, presents double chain DNA and is capable of producing latent infection characterized by low expression of viral genes and minimum cytopathic effects or viral proliferation 21, 22. EBV presents high prevalence in the population, which means that about 90% of adults have antibodies for EBV and normally infect lymphocytes and epithelial cells 21, 22. EBV has been well established as etiological agent of Burkitt's lymphoma and nasopharyngeal carcinoma 21, 22, 23.
EBV normally does not replicate in recently infected lymphocytes B 25. In latent infected lymphocytes B there is expression of six different nuclear proteins or EBNAs, two different membrane proteins or LMP and two filaments of RNA or EBERs 25. These viral products maintain latency and stimulate lymphocytes at rest to continuously proliferate 25. Even though latency means absence of lytic infection, the term is confusing and inappropriate for EBV, since in EBV latent infection is early and there is efficient growth and cell proliferation 25. However, despite the presence of genomes and viral products, proliferation of infected lymphocyte B is similar to the one occurred by the action of the antigen, mytogen or Il 4 25. Regardless, in latency of EBV there is occurrence of proliferation of infected cells and not of viral particles such as it occurs in viral lytic infection 25.
Latency can be directly induced for lytic cycle by activation of BZLF (zebra), a transactivating protein of EBV 25 (Figure 1). Lytic cycle is characterized by intense transcription, replication of DNA and production of late proteins such as capside antigens and glucoproteins 25. Oral pillosa leukoplasia is a disease caused by focal replication, being the only in vivo manifestation of replication infection of EBV 25.
Protein LMP-1 modulates growth and differentiation, induces expression of multiple markers of cell surface, cell activators, antigens and adhesion molecules 23, 24. LMP-1 reduces cell response to differentiation signs, increases invasiveness of the same collagen matrix and can transform human fibroblasts in keratinocytes 22-24. It can induce resistance to apoptosis through activation of anti-apoptotic proteins such as Bcl-2 23, 24. In vitro studies demonstrated that LMP-1 can block apoptosis mediated by p53 in epithelial cells and in Burkitt's lymphoma cells.23,24 The expression of LMP-1 in immunodepressed subjects can induced transformation of lymphocytes B and the onset of lymphoproliferative processes 24.
In addition, it has been associated with a series of malignant neoplasms in general, such as thymic carcinoma, gastric carcinoma, breast cancer and OSCC 21-23. Oncogenic action of EBV seems to involve the latent oncoprotein of membrane 1 (LMP-1).22-24
High prevalence of EBV was demonstrated in OSCC 22. However, prevalence of this association varies a lot according to the geographical region and employed techniques 21, 22. Using PCR, the most sensitive technique, prevalence is about 37.9%, without statistical difference between smoker and non-smoker, alcohol abuser and non-alcohol abuser 21.
A study to assess DNA, RNA and level of EBV protein in OSCC demonstrated high prevalence of PCR in DNA of EBV (100%) 22. However, it did not show, through immunohistochemistry, presence of LMP-1 nor BZLF-1 (zebra), which suggested that EBV did not present transforming potential, owing to absence of LMP-1, nor lytic cycle, by absence of zebra in cases of OSCC 22. This finding suggests that EBV does not present carcinogenic potential in OSCC 22.
Genetic Alterations P53
Another factor normally associated with etiology of OSCC is mutation of p53 20, 26-28. Since the discovery of p53, at the end of the 70's, it was believed that it was responsible for modulation of cell response to endogenous and exogenous stress, such as DNA damage, hypoxia, activation of oncogenesis, viral replication and depletion of rhibonucleotides, being an important gene in tumor suppression 20, 27-30.
P53 can block cell cycle in cell G1 phase with sublethal damage in its genome up to complete repair 27. Moreover, it can induce apoptosis, preventing development of clones of cells with severe damage to DNA 27. The absence of native p53, be it by inactivation or destruction, and some mutations of native proteins are capable of inactivating the function of genome gatekeeper played by p53, allowing survival of cells with damage to DNA (Figure 2) 20, 27, 28.
The association of tobacco and alcohol consumption and mutation of p53 is controversial 26. However, genetic studies demonstrated that the mutated region of the gene is different - in patients with risk factors such as smoking and alcohol abuse, patients without risk factors and patients with lip carcinoma 20, 26. Such mutational differences, in these cases, suggest distinct etiologies, such as exogenous, endogenous stimuli and actions of ultraviolet rays, respectively 20, 26.
High frequency of OSCC occurs in the absence of p53 mutation, in which there seems to be only one inactivation of function, be it by destruction of protein or inactivation of the system, such as HPV infection that causes proteolysis of p53, or caused by overexpression of MDM2 which leads to inactivation of p53 20, 28. All these p53 alterations allow the cell to be released from the mechanism of control of proliferation, performing an important role in cell immortalization 28.
Telomers and Telomerase
Telomers are specialized structures existing in the terminal portion of the chromosome of eukaryotic cells, comprised of repetitions of sequence TTAGG 6,7,29. Telomers protect chromosomes from DNA enzyme rupture, incomplete replication, prevent recombination and aberrant fusions, and ensure complete replication of chromosome during cell division 7, 9, 29. Human telomers of somatic cells suffer progressive shortening during cell division, in which there is loss of terminal sequence of DNA resultant from replication 6, 7, 9, 29. Progressive loss of telomers causes senescence 6, 7, 9, 29.
If there is shortening of telomer, the cell stops dividing 6, 7. Thus, the telomer is recognized as a mitotic clock, being responsible for cell replication capability 6. However, germ cells and stem cells do not suffer telomer shortening since they express telomerase, an enzyme that synthesizes telomeric DNA 7.
Telomerase, rhibonucleoprotein polymerase DNA, contains RNAB component that synthesizes repetition of telomer DNA, compensating the loss of telomers during cell division 29. Theoretically, it is believed that only germ cells have telomerase activity, but recently, some types of cells such as bone marrow cells, gastrointestinal cells and skin basal layer cells presented telomerase activity, even though weak 7.
Absence of telomerase in normal cells leads to progressive erosion of telomer, resulting in incomplete replication, which generates chromosome instability, causing senescence 6. In this case, cell senility occurs because telomers are shortening 7. Too short telomers prevent the cell from replicating, but they also cause chromosome instability and, therefore, tend to suffer mutation 6. Therefore, telomerase would act as a factor that maximizes tumor growth 30.
P53, in turn, interprets shortening of telomer as DNA error, and prevents the cell from replicating and leading to senility status 6. Since there is no telomerase in the cell, it stops to get divided 6. The senile status of the cell is considered a anti-tumor protection mechanism (Figure 3) 6, 28.
Telomerase has been expressed in high percentage in extracts of many different types of cancer 6, 7, 28. It presents expression in 75% of the OSCC, 80% of lung cancer, 84% of prostate cancer, 85% liver, 93% breast cancer, among others 9. Immortalized cell lines, both spontaneously and depending on oncovirus transformation such as HPV, normally are telomerase positive 9. Oncoprotein E6 of high risk HPV is capable of activating telomerase by unknown mechanisms 6, 9, 28. Telomerase activity has been detected also in normal tissues, indicating that telomerase can be a marker of proliferation and not only of carcinogenesis 7, 9, 29.
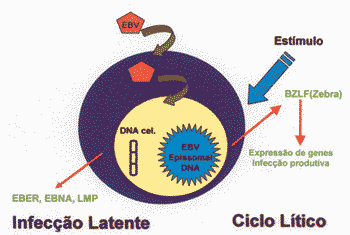
Figure 1. Products of EBV infection in human cells, production of EBER, EBNA and LMPs in latent infection and expression of BZLF-1 (zebra) and LMP-1 in lytic cycle.
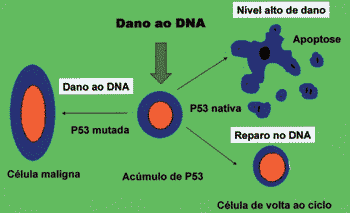
Figure 2. Function of p53 in control of proliferation. Mutated or inactive p53 leads to malignant transformation.
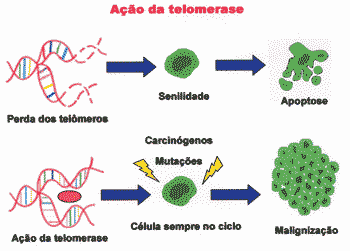
Figure 3. Progressive loss of telomers leads to senility, whereas action of telomerase leads to continuous multiplication, which can result in malignancy.
Reports of the involvement of HPV in oral carcinogenesis are controversial, with infection rates that range from 0 to 87% 15. The results found in the literature, in turn, are difficult to interpret owing to size of samples, methods of tissue collection, preservation of tissue, sensitivity of method, and size of collected tissues. Even though HPV DNA has been detected in OSCC, its etiological role seems to be obscure 2. Previous studies have demonstrated that patients with HPV positive tumors shows greater survival rates than patients with HPV negative tumors 19. Owing to this observation, HPV identification is essential in clinical practice.
A meta-analysis conducted in 1982-1987 indicated that HPV is an independent risk factor and it is extremely important in OSCC, the results indicate that HPV detection is two to three times more common in oral pre-malignant lesions and 4.7 times more common in OSCC, when compared to normal oral mucosa 12. HPV infection seems to be independent from alcohol and tobacco exposure, but it is epidemiologically lower, since HPV infection presents lower prevalence than alcohol and tobacco consumption 12.
Most of the studies do not take into account the age of studied subjects, but there is a study that demonstrated that HPV detection was greater in young male subjects, being more marked in well-differentiated tumors 16. All young non-smokers and non-alcohol abusers presented HPV positive tumors 16. Such results support the theory that in young adults OSCC would be linked to oncogenic virus. In turn, all extremely young patients with OSCC were alcohol abusers and 2 were moderate smokers, and this fact presented importance as the combined effect of HPV and other risk factors, such as alcohol abuse and smoking 16. The top prevalence of HPV in well-differentiated lesions can be determined by viral life cycle, which seems to be better supported by well-differentiated lesions 16.
Some evidence suggests that high risk HPV is completely involved in oral carcinogenesis. HPV has been diagnosed in oral SCC lesions, oral keratinocytes are transformed by high risk HPV via mechanisms that involve E6 and E7, and HPV can be detected both in the primary tumor and in the metastases, being that such results were obtained in studies conducted in geographically different regions 12.
The definition of EBV role in oral carcinogenesis is as uncertain as HPV, and we face the same difficulties concerning the interpretation of studies owing to the same factors related to sample and sensitivity of the method of analysis. However, in EBV cases, the situation is a bit more complicated. EBV infects epithelial cells and lymphocytes, and most analyses do not take into account whether its presence is in neoplastic cells or in inflammatory infiltrate next to the tumor. Moreover, some PCR analyses (the most sensitive method) and exfoliative cytology do not rule out the presence of saliva, which has innumerous lymphocytes. Thus, EBV detection in OSCC ranges from 0 to 100% 21.
In addition, EBV seems to be really implicated in oral carcinogenesis, since the products, such as LMP1, for example, are capable of promoting in vitro malignant transformation 24. Detection of latent EBV infection in pre-malignant lesions and clone expansion caused by EBV suggest that viral infection precedes tumor development, which can be the initial event in oral carcinogenesis 24. Another important finding is the association between presence of EBV and high level of cell atypia, since well-differentiated tumors present smaller number of EBV copies, when compared to little-differentiated tumors 24.
Conversely, EBV can be taken to neoplastic cells by direct contact with infected lymphocytes, being that the infection is a result of low immune status of patients with neoplasia 22. It can be considered quite common, once 90% of the population has EBV antibodies 22.
Once EBV and HPV have products that act directly on cell cycle and apoptosis activation, greater attention should be given to these viruses, since there should be a direct correlation with prognosis. In the EBV case, more studies for the detection of LMP-1 and BZLF-1 are necessary, since such proteins can interact with p53 preventing apoptosis mediated by this route, being that this route is essential for treatment with radiotherapy and chemotherapy, which normally causes apoptosis in malignant cells 23. Thus, the presence of proteins would reduce the efficacy of treatment for OSCC 23. However, only one study was found which correlated EBV and prognosis, but it did not find significant differences between EBV+/; it is important to emphasize that this study detected EBV DNA by PCR, and did not investigate the presence of LMP-1 or BZLF-1 23. For HPV, greater emphasizes should be given to E6 and p53, so that we can be sure that there is influence in treatment and prognosis. It is also importance to point out that we did not find any study that made such correlation in the literature. We can highlight the synergic effect between HPV and EBV, which could act together for oral carcinogenesis.
As a consequence, p53 continues to be the most commonly found mutated gene 20. P53 demonstrates it has a fundamental role in oral carcinogenesis, since it presents frequent mutations and is extremely sensitive to all risk factors, such as HPV, EBV, tobacco, alcohol and telomerase 20. It has an important role in determining prognosis and recurrence, being useful as a diagnostic marker, which can be used even for assessment of tumor margins in local-regional treatment 20. However, many studies have been conducted to assess molecular markers, but most of them had reduced number of samples, and extensive review has indicated that isolated p53 does not have prognostic predictive value, but molecules related to p53 in the control of apoptosis would have a key role in carcinogenesis and should be assessed together 30. In order to elucidate the interaction between p53 and other markers, a comprehensive prospective study would be necessary, in the presence of some etiological agents such as those described above, so that we could assess the role they have in OSCC prognosis.
Telomerase has been described as undetectable in normal cells and tissues, but it is found in germ, immortalized and cancer cells 7. Telomerase expression seems to increase during progression of OSCC, being smaller in dysplasia and higher in frankly invasive lesions, which suggests a close relation between telomerase activity and oral carcinogenesis 29. The findings suggest that telomerase is involved in early stages of oral carcinogenesis 9, 29. Thus, telomerase is genetically activated and represents a critical and initial event, which would lead to carcinogenesis 9, 29.
Conversely, there is a current that does not believe in the previously established model, stating that normal cells would be able to express telomerase in proliferation conditions 9. Therefore, the use of telomerase activity as a marker would generate false positives, depending on the proliferation status under discussion 9. Such findings include the observation that positive telomerase cells present smaller telomers than negative telomerase cells, which seems to suggest that there are other ways of preserving the telomers than telomerase 9. This hypothesis considers that all epithelium of continuous proliferation would be telomerase positive 29. However, there are some factors that should take into account some cells that have the capability to express telomerase, but we can not rule out he possibility that these cells may represent epithelial stem cells 29. Moreover, the presence of telomerase positive cells in normal tissues or areas adjacent to tumors can represent identification of biochemical and molecular events that have not been histologically expressed yet.
We should also analyze here the detection method of telomerase, being that the most frequently used method is TRAP, which means telomeric repetitive amplification protocol. Even though the method is the most sensitive one, it is not specific since it amplifies the whole sequence, and studies demonstrated that the most specific sequence would be in subunits of hTERT.7 Some telomerase subunits are found both in normal cells and in OSCC cells, but only hTERT would be an important marker of differentiation, of potential for cell growth 7. Therefore, there is increase in activity of OSCC telomerase which seems to be related with at least two events in oral carcinogenesis: the first one would be loss of control of stratification and differentiation of epithelium, and the second, individualized overexpression of hTERT in cells.7
Despite the developed understanding of molecular alterations that would lead to malignant transformation, the role of these factors is still obscure. There is wide variety of markers and possible etiological agents currently presented in the scientific literature, which demonstrates the complexity of oral carcinogenesis, a multifactorial event that requires destabilization of many different control systems 30. Even though the studies in this area are exhaustive, incomplete and many times frustrating, they are necessary and should be encouraged, since the best understanding of cell biology and exact knowledge of critical molecular mechanisms to carcinogenesis can lead to directed therapy and more effective management. Moreover, individual variations in the clinical course can be better understood, which would help with predicting prognosis and selecting the appropriate therapy.
CLOSING REMARKSBased on the currently available information, we can state that there are different mechanisms responsible for oral carcinogenesis, from genetic and/or infectious origin, which can range according to geographical, cultural, ethnic and social-economic differences.
Better definition of etiology and cell affections relevant to the evolution of these tumors will allow early diagnosis, appropriate treatment and control and future prevention.
REFERENCES1. Neville BW, Damm DD, Allen CM, Bouquot JE. Patologia Oral e Maxilofacial. 1a ed. Rio de Janeiro: Guanabara-Koogan; 1998. 705p.
2. Gillison ML, Koch WM, Capone RB et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. JNCI 2000; 92(9): 709-20.
3. Llewellyn CD, Johnson NW, Warnakulasuriya KAAS. Risk factors for squamous cell carcinoma of the oral cavity in young people - a comprehensive literature review. Oral Oncol 2001; 37(5): 401-18.
4. Llewellyn CD, Linklater K, Bell J et al. Squamous cell carcinoma of the oral cavity in patients aged 45 years and under: a descriptive analysis of 116 cases diagnosed in the south east of England from 1990 to 1997. Oral Oncol 2003; 39: 106-14.
5. Atula S, Grenman R, Laippala P, Syrjanen S. Cancer of tongue in patients younger than 40 years: a distinct entity? Arch Otolaryngol Head Neck Surg 1996; 122(2): 1313-9.
6. Veldman T, Horikawa I, Barrett JC, Schlegel R. Transcriptional activation of the telomerase hTERT gene by human papillomavirus type 16 E6 oncoprotein. J Virol 2001; 75(9):4467-72.
7. Fujimoto R, Kamata N, Yokoyama K et al. Expression of telomerase components in oral keratinocytes and squamous cell carcinomas. Oral Oncol 2001; 37: 132-40.
8. Scully C. Oral squamous cell carcinoma; from a hypothesis about a virus, to concern about possible sexual transmission. Oral Oncol 2002; 38: 227-34.
9. Belair CD, Yeager TR, Lopea PM, Reznikoff CA. Telomerase activity: a biomarker of cell proliferation, not malignant transformation. Proc Natl Acad Sci USA 1997; 94:13677-82.
10. Lydiatt D. Cancer of the oral cavity and medical malpractice. Laryngoscope 2002; 112(5): 816-9.
11. Oliver RJ, Dearing J, Hindle I. Oral cancer in young adults: report of three cases and review of the literature. Br Dent J 2000; 188(7): 362-6.
12. Miller CS, Johnstone, BM. Human papillomavirus as a risk factor for oral squamous cell carcinoma: a meta-analysis, 1982-1997.Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2001; 91(6): 622-35.
13. Lype EM, Pandey M, Mathew A et al. Oral cancer among patients under the age of 35 years. J Postgrad Med 2001; 47(3):171-6.
14. Wattre P. Apoptose et virus (hormis les retrovirus). Revue Française des Laboratoires 1999; 311: 43-9.
15. Bouda M, Gorgoulis VG, Kastrinakis NG et al. "High risk" HPV types are frequently detected in potentially malignant and malignant oral lesions, but not in normal oral mucosa. Mod Pathol 2000; 13: 644-53.
16. Cruz IBF, Snijders PJF, Steenbergen RDM et al. Age-dependence of human papillomavirus DNA presence in oral squamous cell carcinomas. Oral Oncol 1996; 32B (1): 55-62.
17. Mork J, Lie AK, Glattre E et al. Human papillomavirus infection as a risk factor for squamous cell carcinoma of the head and neck. N Engl J Med 2001; 344(15): 1125-31.
18. Premoli-De-Percoco G, Ramirez JL, Galindo I. Correlation between HPV types associated with oral squamous cell carcinoma and cervicovaginal cytology: an in situ hybridization study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1998; 86(1): 77-81.
19. Capone RB, Pai SI, Koch WM et al. Detection and quantitation of human papillomavirus (HPV) DNA in the sera of patients with HPV - associated head and neck squamous cell carcinoma. Clin Cancer Res 2000; 6(11): 4171-5.
20. Gasco M, Crook T. The p53 network in head and neck cancer. Oral Oncol 2003; 39: 222-31.
21. Sand LP, Jalouli J, Larson P-A, Hirsch J-M. Prevalence of Epstein-Barr virus in oral squamous cell carcinoma, oral lichen planus, and normal oral mucosa. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2002; 93(5): 586-92.
22. Cruz I, Van Den Brule AJ, Brink AA et al. No direct role for Epstein-Barr virus in oral carcinogenesis: a study at the DNA, RNA and protein levels. Int J Cancer 2000; 86: 356-61.
23. Maeda T, Hiranuma H, Matsumura S, Furukawa S, Fuchihata H. Epstein-Barr virus infection and response to radiotherapy in squamous cell carcinoma of the oral cavity. Cancer Letters 1998; 125:25-30.
24. Gonzalez-Moles MA, Gutierrez J, Rodriguez MJ, Ruiz-Avila I, Rodriguez-Archilla A. Epstein-Barr virus latent membrane protein - 1 (LMP-1) expression in oral squamous cell carcinoma. Laryngoscope 2002; 112(3): 482-7.
25. Fields BN, Knipe DM, Howley PM. Fields Virology. Philadelphia: Lippincott-Raven Publishers; 1996.
26. Hsieh LL, Wang P-F, Chen IW et al. Characteristics of mutations in the p53 gene in oral squamous cell carcinoma associated with betel quid chewing and cigarette smoking in Taiwanese. Carcinogenesis 2001; 22(9): 1497-503.
27. Gonzalez-Moles MA, Galindo P, Gutierrez J et al. Expression of the p53 protein in oral squamous cell carcinomas associated with Epstein-Barr virus. Microbios 2000; 102(403):147-54.
28. Optiz OG, Suliman Y, Hahn WC et al. Cyclin D1 overexpression and p53 inactivation immortalize primary oral keratinocytes by a telomerase-independent mechanism. J Clin Invest. 2001; 108(5):725-32.
29. Liao J, Mitsuyasu T, Yamane K, Ohishi M. Telomerase activity in oral and maxillofacial tumors. Oral Oncol 2000; 36: 347-52.
30. Schliephake H. Prognostic relevance of molecular markers of oral cancer - a review. Int J Oral Maxillofac Surg 2003; 32: 233-45.
1 Master studies in Buccodental Pathology under course, UFF, Specialist in Stomatology, UFRJ.
2 Joint Professor, Department of Oral Pathology and Diagnosis, Dental School, UFRJ. Ph.D. in Buccal Pathology, USP.
3 Joint Professor, Department of Pathology, Hospital Universitário Antônio Pedro, Universidade Federal Fluminense. Ph.D. in Buccal Pathology, FOUSP-Bauru.
Department of Pathology, Universidade Federal Fluminense - Niterói / RJ.Post-Graduation Course - Master studies in Buccodental Pathology
Address correspondence to: R. Soldado Francisco de Souza, 110 Rio de Janeiro RJ 22770-155.
Tel (55 21) 3392-0418 - E-mail: beatriz_venturi@ig.com.br
