INTRODUCTIONInner ear malformations affect approximately 20% of all patients with congenital sensorineural hearing loss, and in about 50% of these cases, there is involvement of genetic mechanisms1.
Subotic and Schuster6 (1978) divided congenital sensorineural loss into two large groups: those that comprise bone and membranous labyrinth abnormalities and those that have affections restricted to the individual structures of the membranous labyrinth. In the first group, we may list inner ear congenital malformations and there are innumerous was of classifying them. The reported authors divide them into two types:
1) Complete absence of inner ear (Michel).
2) Cochlear congenital anomalies, depending on the severity of modiolus affection (Mondini).
Ormerod5 divided the malformations into four groups: Michel, Mondini-Alexander, Bing-Sibeman and Scheibe. Schucknecht7 preferred to classify them as Michel, Mondini, Scheibe and Alexander.
As to etiopathogenesis, the malformations may be associated with defects in the induction mechanism of differentiation of neuroectodermic structures of auditory system by chordal mesoderm and neural tube3. Steel et al.4 addressed the possible alterations of neuroepithelial growth process in the genesis of this kind of malformation, based on studies induced in animal models. Unknown genetic factors are probably the key for triggering such abnormalities, although there are also inner ear abnormalities caused by teratogenic agents, such as thalidomide.
The most rare affection in this group is Michel's aplasia. It was described by Michel in 18634, characterized for complete absence of inner ear. However, there were cases reported in the literature of patients classified as Michel's aplasia that differed from the characteristics reported by the author, as confirmed by Ghazli et al.2, who presented two classic bilateral cases of Michel's aplasia in twins.
It is obvious that the affected side has anacusis, but the vestibular function is not affected.
In the present study, we described what we believed to be a classic case of unilateral Michel's aplasia seen in our university hospital.
CASE REPORTIFS, 15 year-old male, came to the Ambulatory of Otorhinolaryngology at Hospital das Clínicas, Universidade Federal de Pernambuco (HC/UFPE), complaining of left hearing loss since birth and left facial paralysis from the age of 3 years and 6 months, during one of the innumerous episodes of otorrhea and otalgia experienced during childhood. The patient did not report problems during gestation, delivery or perinatal period. He did not report diseases during childhood or use of ototoxic drugs. There was no family history of deafness.
Physical examination showed left peripheral facial palsy, House-Brackmann grade V Otoscopy and all ENT examinations were normal. We did not find signs of malformation of any other organ or system. Audiometry showed anacusis on the left and normal hearing on the right.
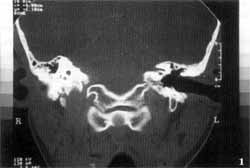
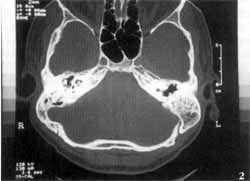
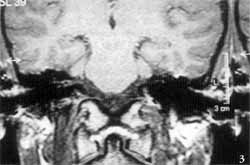
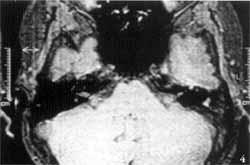
Temporal bone computed tomography (CT scan) showed complete absence of inner ear cavities on the left, in which we could only identify one bone block, corresponding to the otic capsule. External and middle ears, and right temporal bone, were absolutely normal (Figures 1 and 2). We conducted MRI which revealed complete absence of membranous labyrinth, as well as of the VIII cranial nerve (Figures 3 and 4).
The patient and the family were instructed about the nature of the problem and its repercussions on the social and occupational life of patients; we also discussed the need to preserve the normal ear.
DISCUSSIONThe study of inner ear congenital anomalies is confused by the fact that there are different classifications. It is difficult to say if these anomalies belong to groups of distinct etiopathogenesis or if they are simply more or less severe alterations of the same embryonic development of the inner ear, which are named differently because they had been described by different authors. It is probably a fact that the latter holds some truth and that the earlier the etiopathogenic agent affects the inner ear development, the less developed the ear will be at birth. It has been widely studied for Mondini's dysplasia and we are entitled to follow the same reasoning to understand the other abnormalities, including Michel's. However, considering the new and exciting findings about the human genetic code, it is likely that we will determine the genes responsible for one or the other type of malformation in the near future.
The case described by Michel in 1863 was revealed in the autopsy of a 11-year-old deaf child who died in the Hospital of Strasbourg. The most marked characteristic of the case described by the author was the absence of labyrinth and the whole otic capsule, replaced by a thin bony layer that separated the tympanic area from the posterior fossa. The facial nerve was preserved, but VIII cranial nerve did not exist. Alterations were similar on both sides.
It seems that identical cases have only been reported in the study by Ghazli et al.7 The authors believed that the twins described by them were the only cases of classic Michel's syndrome in the literature and that all other reports showed some remains. of the membranous labyrinth, or persistence of otic capsule, despite the absence of membranous structures.
The case we described here fall within this category, because the existence of a compact bony block separating the middle ear from the posterior fossa is very evident, and there is no thin bony layer, as initially described by Michel.
However, we believe that this difference is not very significant for the identification of the anomaly, since we also agree that all these malformations may be represented by the same disease, at different levels of severity. In fact, there is really one key difference among Michel's aplasia and the other forms, which is the complete absence of membranous labyrinth and vestibulocochlear nerve. It may mark an etiopathogenic singularity of Michel's aplasia, because it means, first of all, that the etiological factor has acted before the otic vesicle or even before the auditory placode. Secondly, there is a defect in the induction mechanisms of the neural tube, which is the portion. responsible for the formation of the acoustic nerve from expansions of the notochord.
Concerning Michel's aplasia, another confusing issue is incomplete radiological study. In the past, the absence of high resolution CT scan could have induced diagnostic mistakes, because of paucity of images obtained with the existing methods at that time. Rudimentary inner ear, for example, could have been overlooked.
In addition, CT scan does not provide good visualization of the membranous labyrinth, which is essential to characterize Michel's aplasia. Consequently, MRI is essential to attest the absence of these structures and VIII cranial nerve. In the case reported here, the presence of only one nerve on the superior portion of the rudimentary inner ear canal was evident, and it followed the path of the facial nerve in the temporal bone.
The VIII cranial nerve was clearly absent.
FINAL COMMENTSMichel's aplasia is the most severe form of inner ear malformation, rarely found in its classic form, especially in the detailed format described by the original author. The case reported here includes the main characteristics of this type of malformation, which may be denominated as such. Temporal bone CT scan and MRI are essential tests for precise diagnosis of these anomalies.
Malformation of inner ear is still a fertile ground for etiological investigations. The current lack of definition hinders appropriate classification of the anomalies. Recent progresses in genetic mapping efforts will certainly contribute to a more precise definition.
REFERENCES1. COSTA, S. S.; CRUZ, O. L. M. - Otologia clinica e cirurgica. Revinter. Cap. 4. p. 109-111, 2000.
2. GHAZLI, K.; MERITE-DRANCY, A.; MARSOT DUPUCH, K.; MEYER, B.; JEUNESSE, Y; CHOUARD, C. H. - À propos de deux cas familiaux de syndrome de Michel. Ann. Otolaryngol. Chin Cervicofac., 115: 1, 29-34, 1998.
3: MELNIK, A. R.; WEISS, L. - Mesodermal induction defect as a possible cause of ear malformations. Ann. Otol. Rhinol. Laryngol., 92: 160-4, 1983.
4. MICHEL, E. M. - Memoirs sue less anomalies congenitales de Foreille interne. Gas. Md. Strasburg. 3: 55, 1863.
5. ORMEROD, F. C. - The Pathology of congenital deafness. J. Laryngol. OWL, 74: 919-950, 1960.
6. SUBOTIC, R.; SHUSTER, R. - Simultaneous histological, audiological and radiological findings in congenital anomalies of the inner ear. J. Laryngol. Otol., 92 (4): 281-291, 1978.
7. SCHUKNECHT, H. F. - Mondini dysplasia: a clinical and pathological study. Ann. Otol. Rhinol. Laryngol. (Suppl 65)89: 3-23, 1980.
* Physician, Resident, Discipline of Otorhinolaryngology, Hospital das Clínicas, Universidade Federal de Pernambuco -HC/UFPE.
** Joint Professor, Discipline of Otorhinolaryngology, HC/UFPE.
*** Faculty Professor, Discipline of Otorhinolaryngology, HC/UFPE.
**** Undergraduate, School of Medicine, HC/UFPE.
Affiliation: Universidade Federal de Pernambuco - UFPE - Division of otorhinolaryngology, Hospital das Clínicas da UFPE.
Address correspondence to: Patrícia Ferreira dos Santos - Rua Estrada do Encanamento, 702 - Apto. 401- Bloco C - Casa Forte - 52060-210 Recife/PE.
Tel: (55 81) 3442-5200/(55 81) 9963-9409-E-mail: pfpimentel@elogica.com.br
Article submitted on January 23, 2001. Article accepted on February 6.2001.