INTRODUÇÃOA fibrose cística (FC) é a doença hereditária fatal mais comum entre a população caucasiana
1. Existem aproximadamente 60.000 pessoas com FC em todo o mundo, com incidência estimada em 1:2.906 nascidos vivos brancos e 1:10.338 não brancos nos Estados Unidos. Especificamente, a incidência de FC em hispânicos e afro-americanos é 1:9.200 e 1:15.000 nascidos vivos, respectivamente
2. No Brasil, ainda existem muitas dificuldades para a confirmação diagnóstica desta condição.
Trata-se de disfunção autossômica recessiva, resultante de mutação no gene regulador da condutância transmembrana localizado no cromossomo 7, responsável pelo controle dos processos secretores
3. O defeito genético é carregado por aproximadamente uma em cada 25 pessoas na população e é expresso quando uma criança recebe o gene defeituoso de ambos os pais
4. A mutação ΔF 508 é a mais comum e responde por aproximadamente 66% das mutações em todo o mundo
2.
Doença multissistêmica, caracteriza-se por um amplo espectro de manifestações e complicações resultantes da disfunção generalizada das glândulas exócrinas. Nos pulmões e nas vias digestivas, estas glândulas produzem secreções espessas e viscosas, como consequência do transporte defeituoso do íon cloro através das membranas, levando a obstruções de ductos e destruição tecidual
1.
As complicações clínicas incluem o desenvolvimento de infecções pulmonares supurativas crônicas evoluindo com bronquiectasias, insuficiência pancreática, levando, consequentemente, à síndrome de má-absorção (desnutrição em 85% dos pacientes), diabetes mellitus, doenças hepáticas (principalmente em pacientes mais velhos) e disfunção urogenital (infertilidade masculina). O suor dos pacientes com FC é caracteristicamente salgado. O diagnóstico final é estabelecido por meio da dosagem de cloro e sódio no suor, associada a manifestações clínicas compatíveis
5.
A FC ainda é geralmente citada como uma doença da infância. A morbimortalidade é elevada, porém, ao longo dos anos, o avanço do conhecimento sobre a fisiopatologia e as formas de tratamento tornou cada vez maior a sobrevivência desses pacientes
6. Atualmente, a sobrevida média nos Estados Unidos e na Europa é de aproximadamente 37 anos e mais de 40% das pessoas com FC têm mais de 18 anos
7.
As vias aéreas superiores são comprometidas na quase totalidade dos pacientes na forma de pansinusite crônica. A avaliação tomográfica apresenta-se alterada em quase 100% dos pacientes com FC. No entanto, a grande maioria das crianças não apresenta sintomas de vias aéreas superiores. O diagnóstico de rinossinusite crônica determinado apenas pelos sintomas é subestimado, uma vez que o paciente não valoriza o quadro nasossinusal
8.
Segundo Umetsu et al., o comprometimento nasossinusal poderia exacerbar o quadro pulmonar, já que serviria como fonte de infecção
9.
A polipose nasal ocorre em cerca de 10% das crianças e até 40% dos adultos com FC. O início se dá geralmente por volta dos 8 a 10 anos de idade, sendo incomum em menores de 5 anos. A etiologia é incerta, mas pode estar relacionada a infecções, alergia, fatores imunológicos, alteração das secreções e anormalidades ciliares. Os pólipos são geralmente assintomáticos, mas podem causar obstrução nasal crônica, resultando em respiração oral, cefaleia e alteração do olfato e paladar
10. A prevalência da polipose nasal é aumentada quando o paciente é examinado por meio de nasofibroscopia, ao ser comparado ao exame realizado com o uso de rinoscopia anterior
11.
O presente estudo procura correlacionar a gravidade dos pacientes portadores de fibrose cística com as alterações nasofibroscópicas e tomográficas de seios paranasais. Como objetivos específicos, espera-se: avaliar as manifestações respiratórias de vias aéreas superiores mais comuns nos pacientes com fibrose cística; comparar os achados endoscópicos com os achados de tomografia dos seios paranasais; correlacionar a presença de colonização pulmonar crônica com os achados de nasofibroscopia e tomografia computadorizada de seios paranasais; e correlacionar os achados tomográficos e endoscópicos nasais com o quadro clínico geral dos pacientes por meio do escore clínico de Shwachman-Kulczycki.
MÉTODOEstudo transversal, prospectivo, envolvendo 20 pacientes com diagnóstico de fibrose cística acompanhados no Ambulatório de Pneumologia do Hospital Infantil Albert Sabin (HIAS). O trabalho foi previamente aprovado no Comitê de Ética em Pesquisa do HIAS, sob o protocolo 001/09.
Foram incluídos na pesquisa pacientes entre 6 e 25 anos de idade, de ambos os sexos, com diagnóstico comprovado de FC, evidenciado por dois testes de dosagem de cloro e sódio no suor positivos ou presença de mutações FC conhecidas identificadas por teste genético. Todas as crianças incluídas no estudo tiveram termo de consentimento assinado por seus responsáveis. Os maiores de 18 anos assinaram de próprio punho esse termo.
Foram excluídos pacientes com evidências clínicas e/ou radiológicas de agudização de doença pulmonar e/ou nasossinusal no momento da realização do estudo, assim como todos aqueles cujos responsáveis não consentiram sua participação.
Os pacientes foram avaliados com o uso de nasofibroscopia, a qual foi realizada no Ambulatório de Otorrinolaringologia do Hospital Universitário Walter Cantídio da Universidade Federal do Ceará (HUWC-UFC), utilizando fibra óptica flexível de 3,4 mm de diâmetro, da marca Olympus BF type 3C40 (
Olympus Corporation, Tóquio, Japão) ou fibra óptica rígida de 0º e 4 mm de diâmetro, da marca Asap (
Asap Endoscopic Products Gmgh, Freiburg, Alemanha). Previamente ao início do exame, procedeu-se à aplicação de spray nasal de solução vasoconstrictora a base de cloridrato de nafazolina 0,05%, associado ao anestésico neotutocaína 2%, com a finalidade de facilitar a passagem da fibra óptica e diminuir o desconforto do paciente. Por meio deste exame, realizou-se uma avaliação direta da cavidade nasal, como uma forma de identificar a presença de alterações anatômicas no septo nasal e/ou nas estruturas da parede nasal lateral, especialmente a presença de polipose nasossinusal. Os achados nasofibroscópicos foram classificados de acordo com os critérios de Meltzer
12 (Quadro 1).
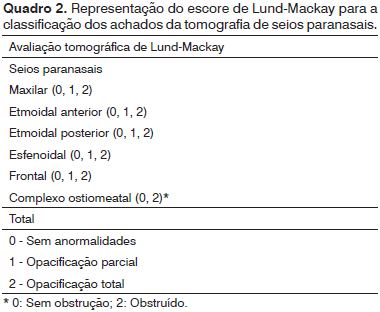
Foram feitas, ainda, tomografias de seios paranasais no serviço de radiologia do Hospital Infantil Albert Sabin, em um período de no máximo 30 dias após realização da nasofibroscopia. Foram feitas em planos coronais e axiais com cortes de 2,0 e 3,0 mm de espessura, com técnica de baixa dose (130 KV e 40 mA). Não houve necessidade de sedação nesses pacientes. Os achados tomográficos foram classificados de acordo com os critérios de Lund-Mackay
13 (Quadro 2). Só foram pontuados os seios pneumatizados. Para possibilitar a comparação de resultados, fez-se a correção para o escore original de 0-24. Portanto, multiplicou-se o valor obtido por 24/(total de seios da face pneumatizados)
8.
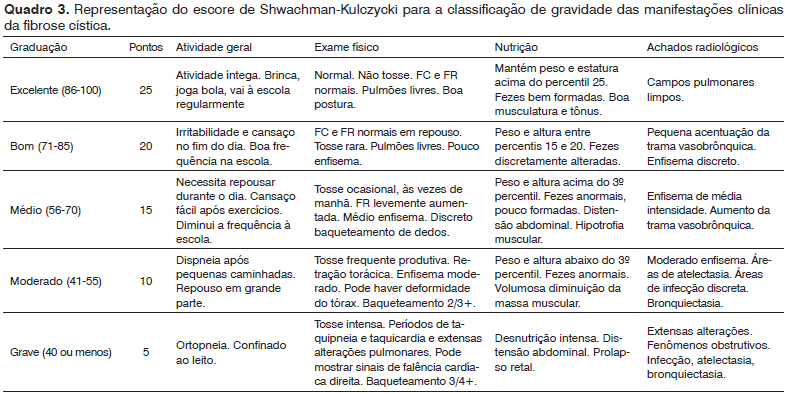
A avaliação clínica para determinar a gravidade das manifestações da fibrose cística nesses pacientes foi realizada com o uso do escore clínico de Shwachman-Kulczycki
14 (Quadro 3).
Para processar os dados, elaborar gráficos e auxiliar as análises estatísticas, utilizou-se o programa
GraphPad Prism versão 4.00. O teste de Spearman foi utilizado para estudar a correlação entre os achados tomográficos e nasofibroscópicos e entre o escore de Shwachman e achados tomográficos e nasofibroscópicos. O teste exato de Fisher foi utilizado para estudar a correlação entre a presença de colonização pulmonar crônica e os achados tomográficos e nasofibroscópicos. Considerou-se como estatisticamente significativo o valor de
p < 0,05.
RESULTADOSDos 20 pacientes selecionados a participar do estudo, oito (40%) eram do sexo feminino e 12 (60%) do sexo masculino e a idade variou de 6 a 25 anos (média 12,6 ± 5,73) (Figura 1).
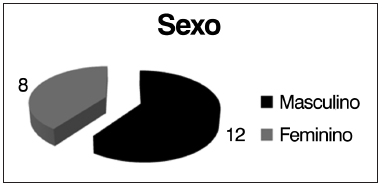
Figura 1. Distribuição quanto ao sexo.
As manifestações de vias aéreas mais comuns nos pacientes avaliados foram tosse crônica (65%), obstrução nasal (45%), cefaleia (35%), rinorreia/descarga pós-nasal (30%) e dor facial (20%) (Figura 2).
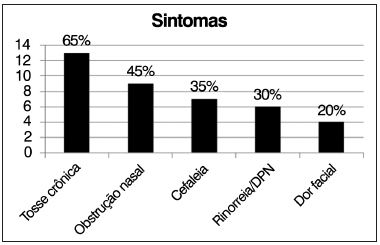
Figura 2. Distribuição de frequências dos sintomas respiratórios.
Dos 20 pacientes selecionados, 16 foram avaliados com cultura de lavado broncoalveolar para determinar a presença de colonização pulmonar crônica. Destes, 15 pacientes (75%) eram colonizados crônicos por
Pseudomonas aeruginosa. Dentre esses, seis (30%) eram também colonizados por
Staphylococcus aureus e um (5%) apresentava ainda
Burkholderia cepacea (Figura 3).
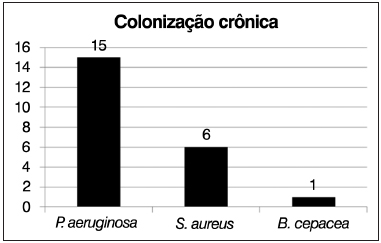
Figura 3. Representação da presença de colonização pulmonar crônica.
A presença de alterações nas imagens de tomografia computadorizada de seios paranasais, de acordo com os critérios de Lund-Mackay, foi observada em 94,1% dos 17 pacientes avaliados (três pacientes não realizaram tomografia: dois foram a óbito antes da realização do exame por agudização do quadro pulmonar e um não compareceu ao agendamento), enquanto apenas 5,9% apresentavam tomografia de seios paranasais normal (Figura 4).
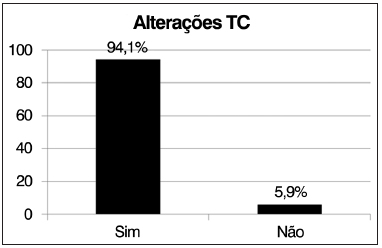
Figura 4. Frequência de alterações observadas à tomografia de seios paranasais.
Dos pacientes avaliados, dez (50%) apresentavam alterações nasofibroscópicas de acordo com a classificação de Meltzer, sendo as alterações mais comuns edema de meato médio em sete (35%) pacientes, pólipos em meato médio em três (15%) e descarga de secreção purulenta de meato médio em um (5%) paciente (Figura 5).
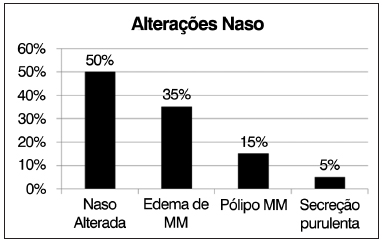
Figura 5. Distribuição de frequências de alterações observadas à nasofibroscopia.
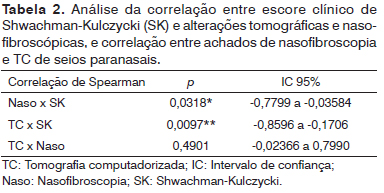
Não se observou correlação estatisticamente significante entre a intensidade dos achados de nasofibroscopia e de TC de seios paranasais nos pacientes avaliados (
p = 0,4901). Entretanto, observamos correlação inversa entre o valor do escore clínico de Shwachman-Kulczycki e os achados de nasofibroscopia (
p = 0,0318) e de TC de seios paranasais (
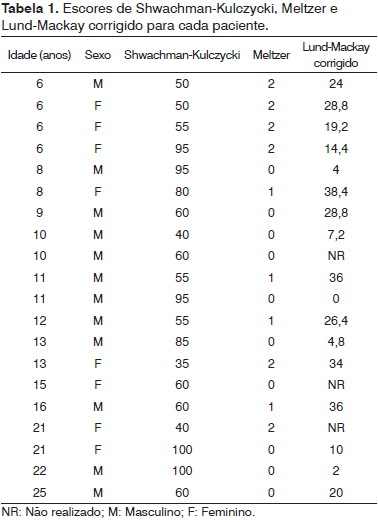
p = 0,0097), o que significa que os pacientes clinicamente mais graves apresentaram mais alterações na nasofibroscopia e TC de seios paranasais (Tabelas 1 e 2).
Foi observada correlação estatisticamente significante entre a presença de colonização pulmonar crônica e alterações nasofibroscópicas (
p = 0,0325), ou seja, pacientes colonizados crônicos apresentaram maior escore nas alterações de nasofibroscopia (Figura 6). Porém, esta correlação não foi observada com as alterações na TC de seios paranasais (
p = 0,2941), ou seja, independentemente da presença de colonização crônica, a maioria dos pacientes apresentou alterações tomográficas (Figura 7).
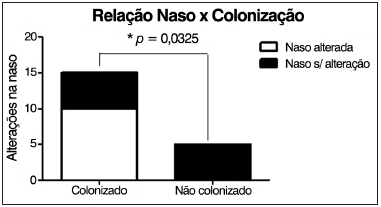
Figura 6. Correlação entre a presença de colonização pulmonar crônica e alterações nasofibroscópicas.
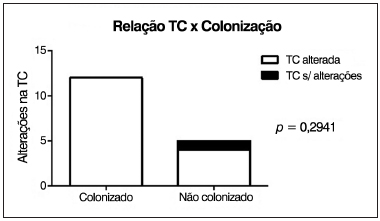
Figura 7. Correlação entre a presença de colonização pulmonar crônica e alterações na TC de seios paranasais.
O seguimento dos pacientes com fibrose cística (FC) tem requerido, dentro da abordagem multidisciplinar, uma atenção cada vez maior dos otorrinolaringologistas, uma vez que diversos estudos têm sido desenvolvidos para analisar o impacto da rinossinusite crônica na função pulmonar dos pacientes acometidos, o que pode aumentar substancialmente a morbimortalidade destes
11. Por outro lado, os avanços terapêuticos têm aumentado a sobrevida destes pacientes, possibilitando a inclusão de crianças e adultos em estudos científicos que, tais como este, abordam essa afecção.
Os pacientes avaliados em nosso estudo apresentaram elevada prevalência de sintomas nasossinusais, como obstrução nasal, cefaleia, rinorreia/descarga pós-nasal e dor facial, o que é discordante com o que foi descrito por Boari & Castro Jr
8, os quais demonstraram que a prevalência dos sintomas nasossinusais nos pacientes portadores de FC é muito baixa, justificando que isso se deve, provavelmente, à subvalorização destes sintomas em detrimento de manifestações clínicas mais graves e que afetam com maior intensidade sua qualidade de vida.
Dos 20 pacientes selecionados, quinze (75%) eram colonizados crônicos por
Pseudomonas aeruginosa; dentre esses, seis (30%) eram também colonizados por
Staphylococcus aureus e um (5%) apresentava ainda
Burkholderia cepacea. Esses dados são concordantes com o que é apresentado na literatura, já que esses patógenos são os mais frequentemente descritos nas publicações como agentes causais de infecção pulmonar crônica em pacientes portadores de FC
3.
A presença de alterações nas imagens de tomografia computadorizada (TC) de seios paranasais, de acordo com os critérios de Lund-Mackay, foram observadas em 94,1% dos 18 pacientes avaliados, dado compatível com as descrições de Henriksson et al., que evidenciam que praticamente 100% dos portadores de FC apresentam alterações tomográficas nasossinusais
8,11.
Dez (50%) dos pacientes avaliados apresentavam alterações nasofibroscópicas, de acordo com a classificação de Meltzer, sendo compatível com a publicação de Umetsu et al.
9, que demonstram que a análise nasofibroscópica é a de maior sensibilidade para avaliar a presença de achados compatíveis com rinossinusite nesses pacientes. Além disso, ainda na avaliação nasofibroscópica, 15% dos pacientes apresentavam pólipos nasais, o que também é compatível com a literatura, que descreve que a polipose nasal ocorre em cerca de 10% das crianças com FC
10.
Foi observada correlação estatisticamente significativa entre a presença de colonização pulmonar crônica e alterações nasofibroscópicas (
p = 0,0325), porém, esta correlação não foi observada com as alterações na TC de seios paranasais (
p = 0,2941). Não foram identificados na literatura consultada trabalhos que demonstrem e/ou comprovem essa correlação.
Não se observou correlação estatisticamente significante entre a intensidade dos achados de nasofibroscopia e de TC de seios paranasais nos pacientes avaliados (
p = 0,4901), discordando do que foi descrito por Umetsu et al.
9, os quais observaram correlação estatisticamente significante entre achados nasofibroscópicos e tomográficos em pacientes com FC. Entretanto, observou-se correlação inversa entre o valor do escore clínico de Shwachman-Kulczycki e os achados de nasofibroscopia (
p = 0,0318) e de TC de seios paranasais (
p = 0,0097), o que significa que os pacientes clinicamente mais graves apresentaram mais alterações na nasofibroscopia e TC de seios paranasais.
CONCLUSÃOOs sintomas respiratórios mais comuns nos pacientes avaliados foram tosse crônica, obstrução nasal, cefaleia, rinorreia e dor facial.
Não houve correlação entre a intensidade das alterações tomográficas e endoscópicas nasais.
Houve correlação direta entre a presença de colonização crônica e os achados nasofibroscópicos, porém, não houve correlação com os achados tomográficos.
Houve correlação inversa entre o escore clínico de Shwachman-Kulczycki e os achados tomográficos e de nasofibroscopia.
REFERÊNCIAS 1. MacLusky I, Levison H. Cystic Fibrosis. In: Chernick V, Boat F, eds. Kendig's Disorders of the Respiratory Tract in Children. Philadelphia: WB Saunders; 1998. p.838-82.
2. Anselmo MA, Lands LC. Cystic Fibrosis. In: Taussig LM, Landau LI, eds. Pediatric Respiratory Medicine. Philadelphia: Mosby Elsevier; 2008. p.845-57.
http://dx.doi.org/10.1016/B978-0323040488.50064-5 3. Koch C. Early infection and progression of cystic fibrosis lung disease. Pediatr Pulmonol. 2002;34(3):232-6.
http://dx.doi.org/10.1002/ppul.10135 4. Dodge JA, Morison S, Lewis PA, Colest EC, Geddes D, Russell G, et al. Cystic fibrosis in the United Kingdom, 1968-1988: incidence, population and survival. Paediatr Perinat Epidemiol. 1993;7(2):157-66.
http://dx.doi.org/10.1111/j.1365-3016.1993.tb00390.x 5. Boat TF. Cystic Fibrosis. In: Murray JF, Nadel JA, eds. Textbook of respiratory medicine. Philadelphia: WB Saunders; 1988. p.1126-52.
6. Dalcin Pde T, Abreu e Silva F. Fibrose cística no adulto: aspectos diagnósticos e terapêuticos. J Bras Pneumol. 2008;34(2):107-17.
7. Yankaskas JR, Marshall BC, Sufian B, Simon RH, Rodman D. Cystic fibrosis adult care: consensus conference report. Chest. 2004;125(1 Suppl):1S-39S.
http://dx.doi.org/10.1378/chest.125.1_suppl.1S 8. Boari L, Castro Jr NP. Diagnosis of chronic rhinosinusitis in patients with cystic fibrosis: correlation between anamnesis, nasal endoscopy and computed tomography. Braz J Otorhinolaryngol. 2005;71(6):705-10.
9. Umetsu DT, Moss RB, King VV, Lewiston NJ. Sinus disease in patients with severe cystic fibrosis: relation to pulmonary exacerbation. Lancet. 1990;335(8697):1077-8.
http://dx.doi.org/10.1016/0140-6736(90)92642-U 10. Clinical guidelines: Care of children with cystic fibrosis. Royal Brompton Hospital. 2007 4th edition. Available from
http://www.rbht.nhs.uk/childrencf 11. Henriksson G, Westrin KM, Karpati F, Wikström AC, Stierna P, Hjelte L. Nasal polyps in cystic fibrosis: clinical endoscopic study with nasal lavage fluid analysis. Chest. 2002;121(1):40-7.
http://dx.doi.org/10.1378/chest.121.1.40 12. Meltzer EO, Hamilos DL, Hadley JA, Lanza DC, Marple BF, Nicklas RA, et al.; Rhinosinusitis Initiative. Rhinosinusitis: Developing guidance for clinical trials. Otolaryngol Head Neck Surg. 2006;135(5 Suppl):S31-80.
http://dx.doi.org/10.1016/j.otohns.2006.09.01413. Lund VJ, Mackay IS. Staging in rhinosinusitus. Rhinology. 1993;31(4):183-4.
14. Schwachman H, Kulczycki LL. Long-term study of one hundred five patients with cystic fibrosis; studies made over a five- to fourteen-year period. AMA J Dis Child. 1958;96(1):6-15.
1. Pós-Doutor (Professor Adjunto de Otorrinolaringologia da Faculdade de Medicina da Universidade Federal do Ceará).
2. Especialista (Médica Otorrinolaringologista).
3. Especialista (Médica Assistente do Serviço de Pneumologia Pediátrica do Hospital Infantil Albert Sabin).
4. Especialista (Médico Assistente do Serviço de Radiologia do Hospital Infantil Albert Sabin).
5. Doutorado (Professora Associada da Disciplina de Pneumologia da Faculdade de Medicina da Universidade Federal do Ceará. Médica Assistente do Serviço de Pneumologia Pediátrica do Hospital Infantil Albert Sabin).
Faculdade de Medicina da Universidade Federal do Ceará e Hospital Infantil Albert Sabin.
Endereço para correspondência:
Marcos Rabelo de Freitas
Departamento de Cirurgia, Faculdade de Medicina da Universidade Federal do Ceará
Rua Profesor Costa Mendes, nº 1608, Rodolfo Teófilo
Fortaleza - CE. Brasil. CEP: 60416-160
Tel: (85) 3366-8062
E-mail:
marcosrabeloufc@gmail.com Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) do BJORL em 18 de janeiro de 2013. cod. 10717.
Artigo aceito em 15 de maio de 2013.