INTRODUÇÃOA síndrome de Peutz-Jeghers foi descrita pela primeira vez em 1896 por um cirurgião londrino, Jonathan Hutchinson1. Todavia, a associação entre hereditariedade na síndrome foi primeiramente reconhecida por Peutz (1921), numa família alemã, cujas características clínicas incluíram polipose gastrintestinal, pigmentação mucocutânea, polipose nasal e extrusão retal de pólipos2. Em 1949, os médicos Jeghers, Mukusiak e Katz perceberam a presença de herança autossômica dominante tornando a síndrome melhor conhecida na comunidade científica3. A SPJ é identificada a partir da tríade pigmentação da pele e de membranas mucosas, polipose hamartomatosa do trato gastrointestinal e tendência familiar1,4. A polipose nasal foi mencionada por Peutz em sua observação inicial e desde então foi poucas vezes citada na literatura especializada5,6,7,8,9.
Publicações dos últimos anos descrevem uma maior morbi-mortalidade relacionada à SPJ referente às complicações da própria doença no trato gastrintestinal6,10,11,12 ou à maior incidência de câncer em diferentes sistemas6,9. Os avanços das pesquisas genéticas permitiram a identificação dos gens envolvidos nas mutações cromossômicas responsáveis pela malignização dos pólipos e melanose13,14,15.
Recentemente, presenciamos um caso de polipose nasal bilateral, de nasofaringe e seios maxilares em menino de 14 anos de idade, com o diagnóstico clínico de sinusite crônica e passado mórbido de tratamento cirúrgico para obstrução intestinal por múltiplos pólipos. O objetivo desse trabalho é mostrar uma associação rara em ORL, a SPJ, chamando a atenção de cirurgiões, pediatras e especialistas para a importância do diagnóstico da polipose nasossinusal.
APRESENTAÇÃO DE CASO CLÍNICOCriança de quatorze anos de idade, sexo masculino, raça negra, foi submetida há seis anos à ressecção jejuno-ileal devido à intussuscepção e necrose de alça intestinal. No estudo anátomo-patológico da peça cirúrgica foram encontrados múltiplos pólipos que microscopicamente foram classificados como hamartomas. Apresentava ao exame físico importante pigmentação labial, peri-oral (Figura 1) e de polpas digitais (Figura 2). Como havia, também, alterações tegumentares em irmã mais nova e pai, impôs-se o diagnóstico de síndrome de Peutz-Jeghers.
Dois anos após, o paciente apresentou quadro de obstrução nasal, coriza hialina e ronco noturno. Procurou o pediatra que diagnosticou rinite alérgica, iniciando tratamento clínico, sem melhora dos sintomas. A partir de então, apresentou vários episódios de febre, rinorréia purulenta, cacosmia, além de dificuldade respiratória contínua. Seguido pela disciplina de Pediatria, foi tratado durante um ano de sinusite crônica. Sem resultado, foi encaminhado à disciplina de ORL para melhor investigação. Na ectoscopia, observou-se edema frontal e alargamento de dorso e ponta nasais. A rinoscopia anterior mostrou uma grande massa acinzentada preenchendo a cavidade nasal, pouco depressível, bilateralmente e com secreção purulenta fétida. Os exames laboratoriais de rotina não mostraram anormalidades. As radiografias de seios paranasais em diferentes incidências evidenciaram velamento de ambos os seios maxilares (Figura 3). A tomografia computadorizada não conseguiu identificar os cornetos e mostrou polipose nasal bilateral com velamento de seios paranasais (Figura 4).
Com base nesses resultados, programou-se a ressecção cirúrgica desses pólipos (Caldwell-Luc). Durante o ato cirúrgico, foram observados múltiplos pólipos em nasofaringe, fossas nasais e seios maxilares, de coloração acinzentada, consistência aumentada, importante mucocele maxilar bilateral e destruição das paredes laterais do nariz. Ressecou-se todo o material da nasofaringe e dos seios maxilares, enviando-o para o estudo histopatológico. Após a cirurgia, foram prescritas limpeza local com soro fisiológico 0,9% e rifocina tópica.
Exame histopatológico
Macroscopia
Em fossas nasais e cavum: múltiplas formações polipóides branco-pardacentas, pesando 120g e medindo em conjunto 8,0x7,5x3,0cm, de consistência endurecida.
Microscopia
Pólipos revestidos por epitélio do tipo respiratório, exibindo áreas de metaplasia escamosa madura e imatura. A submucosa apresenta intenso edema e infiltrado inflamatório constituído predominantemente por plasmócitos e linfócitos e por ocasionais eosinófilos. Notam-se ainda dilatações císticas preenchidas por muco e focos de infiltrado inflamatório neutrofílico que permeiam o epitélio.
O paciente vem sendo acompanhado clinicamente há dez meses e não apresenta recidiva dos pólipos (Figura 5). No momento, permanece sem queixas, com fisiologia normal das vias aéreas superiores.
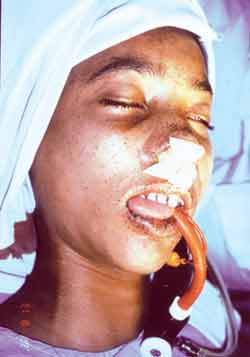
Figura 1. Pigmentação muco-cutânea em lábios e região peri-oral de paciente com a Síndrome de Peutz-Jeghers.
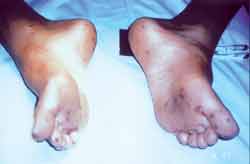
Figura 2. Pigmentação muco-cutânea em região plantar de paciente com a Síndrome de Peutz-Jeghers.
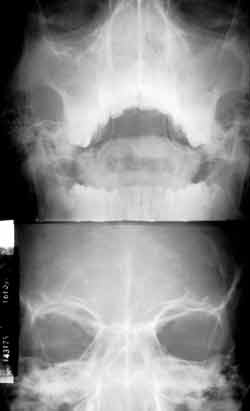
Figura 3. RX dos seios da face (MNP e FNP): velamento de seios paranasais.
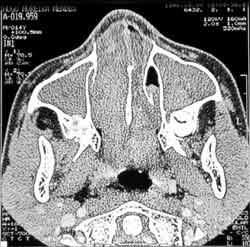
Figura 4: Tomografia computadorizada: polipose nasal bilateral com velamento dos seios paranasais, não sendo possível a identificação dos cornetos.
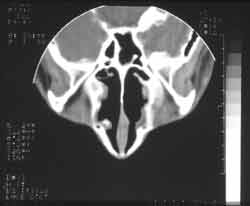
Figura 5: Controle pós-operatório: paciente sem queixas, fisiologia normal das vias aéreas superiores, com acompanhamento anual para detecção das recidivas.
Trata-se do relato de uma doença rara (SPJ) associada com polipose nasal em criança de 14 anos de idade. O diagnóstico do paciente foi baseado nos antecedentes patológicos, pessoais e familiares e no exame histopatológico do material colhido na cirurgia nasossinusal, conforme os relatos da literatura4,3,2.
A pigmentação acomete principalmente lábios e mucosa oral (94%)1,3, mãos (74%), pés (62%) ou qualquer outra região do corpo (21%)12. O nosso paciente apresentava típica pigmentação labial, peri-oral e de mãos e pés. A polipose pode ocorrer em qualquer região do trato gastrintestinal, porém é mais comum no jejuno4, a qual também foi observada em nosso paciente. Os pacientes não se tornam sintomáticos até a segunda década de vida11,2, quando os pólipos tendem a infartar, ulcerar, sangrar ou serem a causa de intussuscepção e obstrução intestinal6,12. Com oito anos de idade, a criança desse relato foi submetida à ressecção jejuno-ileal devido à intussuscepção e necrose de alça intestinal. No estudo da peça cirúrgica foram encontrados múltiplos pólipos, classificados microscopicamente como hamartomas. Mais raramente, os pólipos foram encontrados na boca, esôfago, ureter, bexiga, pelve renal, brônquios, nariz, seios maxilares e pulmões4. O nosso paciente apresentava polipose nasossinusal bilateral, com importante repercussão clínica. A confirmação de história familiar da SPJ contribui para o diagnóstico, porém, obter esse dado em nossa sociedade é uma tarefa árdua. No presente caso, a investigação dos parentes mais próximos da criança obteve dados muito sugestivos da síndrome em pai e irmã mais nova: pigmentação típica e queixas que justificaram a investigação. O gen de susceptibilidade da SPJ codificado por STK11 (expressa em serino/theorinine quinase, também chamado LKB1), foi identificado em famílias com a síndrome. Resultados confirmam o mapeamento do locus comum no 19p13.3 mas também sugere a existência na minoria das famílias de um locus potencial, no 19q13.413.
A sobrevivência das famílias é diminuída freqüentemente por emergências cirúrgicas como as complicações dos pólipos que geram operações repetidas, podendo resultar na Síndrome do Intestino Curto10. Além disso, a SPJ associa-se a câncer de pulmão, tumores do cordão sexual ovariano, adenoma maligno (uma forma maligna de câncer cervical) e tumores testiculares feminizantes de células de Sertolli em meninos pré-puberes6,9. A identificação de mutações da linha germinativa nessas famílias poderia ser o ponto inicial do manuseio da síndrome, pois contribui para o desenvolvimento de ambas formas esporádica e familial de câncer14,15, e de doenças malignas em trato gastrintestinal e tecidos extraintestinais9.
Apesar de Peutz incluir a polipose nasal em sua definição da síndrome, ela tem sido descartada, pois se pensava que a polipose nasal ocorria tão comumente na população geral que não poderia ser específica da síndrome9. Westerman et al. apresentaram seis casos de polipose nasal encontrada nos descendentes da família original investigada por Peutz9. Um desses desenvolveu carcinoma de células escamosas da cavidade nasal. Não estava claro se a polipose nasal naqueles pacientes, classificados como pólipos inflamatórios, poderia ser considerada como hamartomas da SPJ. A associação da polipose nasal e SPJ também foi relatada por outros autores5,6,7,8. Cerqua e colaboradores lutaram pela redefinição da síndrome para incluir a polipose nasal5. O desenvolvimento de câncer da cavidade nasal em um paciente com SPJ e com polipose nasal não havia sido relatado antes do relato de Westerman, apesar de De Facq relatar transformação adenomatosa em dois pólipos nasais em um paciente com SPJ6.
COMENTÁRIOS FINAISCom base no que foi exposto, lembramos aos otorrinolaringologistas, pediatras e cirurgiões que a SPJ constitui doença com importante morbi-mortalidade e sugerimos que a polipose nasal faça parte de seu quadro clínico. Assim sendo, essa síndrome deveria ser investigada em pacientes pediátricos que possuam alguma característica típica associada a distúrbios de vias aéreas superiores, que não respondam à terapêutica correta. Dessa forma, conseguiríamos realizar o diagnóstico mais precocemente, contribuindo para a melhor sobrevivência das famílias acometidas. Insistimos que novos relatos devam ser feitos para maior compreensão dessa doença rara com associação em ORL.
REFERÊNCIAS BIBLIOGRÁFICAS1. Hutchinson J. Pigmentation of lips and mouth. Arch Surg 1896;7: 290.
2. Peutz JL. A very remarkable case of familial polyposis of the mucous membrane of the intestinal tract and nasopharynx accompanied by peculiar pigmentations of the skin and mucous membrane. Ned Tijdschr Geneeskd 1921;10:134-146.
3. Jeghers H, Mckusick VA, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits. New Engl JL 1949;241:992-1005.
4. Hemminki A. The molecular basis and clinical aspects of Peutz_Jeghers syndrome. Cell Mol Life Sci 1999;55:735-750.
5. Cerqua N, D'ottavi LR, Perotti V. Rare manifestazioni dell poliposi nasale nella sindrome di Peutz-Jeghers. Acta Otorhinol Ital 1993;13:333-338.
6. De Facq L, De Sutter J, De Man M et al. A case of Peutz-Jeghers syndrome with nasal polyposis, extreme iron deficiency anemia, and hamartoma-adenoma transformation: management by combined surgical and endoscopic approach. Am J Gastroenterol 1995;90:1130-1132.
7. Jancu J. Peutz-Jeghers syndrome involvement of the gastrointestinal and upper respiratory tracts. Am J Gastroenterol 1971;56: 545-549.
8. Manegold BC, Bussmann JF Fürstenberg HS. Klinischer beitrag zum Peutz-Jeghers-syndrom mit befall des magendarmtraktes, der oberen luftwege sowvie beider mammae. Med Welt 1969;25:1435-1439.
9. Westerman AM, Entius MM, De Baar E et al. Peutz-Jeghers syndrome: 78-year follow-up of the original family. Lancet 1999;353:1211-5.
10. Choi HS, Park YJ, Park JG. Peutz-Jeghers syndrome: a new understanding. J korean Med Sci 1999;14:2-7.
11. Foley TR, Garrity TJ, Abt AB. Peutz-Jeghers syndrome: A clinicopathologic survey of the "Harrisburg Family"with a 49-year follow up. Gastroenterol 1988;95:1535-1540.
12. Utsunomiya J, Gocho H, Miyanaga T et al. Peutz-Jeghers syndrome: Its natural course and management. John Hopkins Med J 1975;136:71-82.
13. Mehenny H, Blouin JL, Radhakrishna U et al. Peutz-jeghers syndrome: confirmation of linkage to chromosome 19p13.3 and identification of a potential second locus, on 19q13.4. Am J Hum Genet 1997;61:1327-34.
14. Su GH, Hruban RH, Bansal RK et al. Germiline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am J Pathol 1999;154:1835-40.
15. Trojan J, Brieger A, Raedle J et al. Peutz Jeghers syndrome: molecular analysis of a three-generation kindred with a novel defect in the serine threonine kinase gene STK11. Am J Gastroenterol 1999;94:257-61.
1 Professora Adjunta Mestre-Doutora da Disciplina de Cirurgia Pediátrica da Faculdade de Medicina do Triângulo Mineiro (FMTM).
2 Acadêmicos de Medicina da FMTM.
3 Cirurgiã Pediátrica.
4 Médica Otorrinolaringologista.
Endereço para correspondência: Dra. Adriana Cartafina Perez Bóscollo - Disciplina de Cirurgia Pediátrica - Departamento de Cirurgia - Hospital Escola
Faculdade de Medicina do Triângulo Mineiro
Rua Getúlio Guaritá nº 130 - Bairro Abadia, Uberaba, MG, Brasil 38025-440 - Fone: (0xx34)318.5228 - Fax: (0xx34)312.2390 - E-mail: boscollo@mednet.com.br
Trabalho apresentado no XXI Congresso Brasileiro de Cirurgia Pediátrica, outubro de 2000 - Porto Alegre - RS - Brasil.
Artigo recebido em 11 de setembro de 2000. Artigo aceito em 15 de agosto de 2001.
