INTRODUÇÃOA surdez é a deficiência sensorial mais frequente em seres humanos, apresentando incidências que variam de 1 em 300 a 1 em 1000 crianças em todo mundo1-3. No Brasil essa frequência é estimada em quatro a cada 1000 nascimentos4. Aproximadamente metade dos casos em todo mundo apresenta etiologia genética, incluindo as formas sindrômicas e não-sindrômicas. A surdez não-sindrômica é responsável por 60-70% dos casos da surdez hereditária envolvendo mais de 100 genes diferentes com padrões de herança autossômica dominante (DFNA), autossômica recessiva (DFNB), ligada ao X (DFN) e herança mitocondrial5, entretanto, a herança autossômica recessiva é mais comum. Para muitas populações, a maior causa da surdez não-sindrômica de herança autossômica recessiva é consequência da alteração da proteína Conexina 26, uma proteína de junção comunicante codificada pelo gene GJB2 (13q11-12)(OMIM 121011)6-13.
As conexinas são proteínas transmembranas que formam estruturas hexaméricas cilíndricas de superfície celular tais estruturas se ligam a outros hexâmeros de conexinas em células adjacentes, formando os canais de comunicação intercelular14,15. Na orelha interna a conexina 26 pode estar associada com outras conexinas. A conexina 26 recicla os íons potássio como parte de um mecanismo de transdução de sinal na orelha interna16.
Mutações em três genes codificadores de conexinas, GJB2(Cx 26), GJB6(Cx 30), e GJB3(Cx 31) foram identificados como causadores de deficiência auditiva15,16.
Dentre as mutações no gene GJB2 que ocasionam DFNB descritas até o momento, a mutação 35delG é responsável pela maioria dos alelos mutantes (60-85%) na população caucasiana6,7,9-13,17.
A mutação 35delG é a deleção de uma base guanina em uma sequência de seis guaninas, que se estendem da posição 30 a 35 dos nucleotídeos, no éxon codificante do gene GJB2, resultando na formação de um códon de terminação. Esta deleção resulta na síntese de um polipeptídio incompleto, com 12 aminoácidos, diferentemente do normal com 226 aminoácidos18.
Análises do gene GJB2 em pacientes com surdez de herança AR, especialmente da mutação 35delG, revelaram que cerca de 10 a 50% dos pacientes eram portadores de somente 1 alelo mutante19. Estudos posteriores sugeriram a existência de outra mutação no éxon codificante do gene da Cx 26, ou a possível coexistência da mutação del (GJB6-D13S1830), no gene da Cx30, nos indivíduos heterozigotos para o gene GJB220-22.
No Brasil cerca de 80% dos casos de surdez são causados por fatores ambientais, e os 20% restantes devem ser causados por fatores hereditários4. Um estudo recente realizado no estado de São Paulo mostrou que a mutação 35delG foi a mais frequente entre os pacientes analisados (12,4%), presente em 23% dos casos familiais e 6,2% dos casos esporádicos. A segunda mutação mais frequente encontrada pelos autores foi a deleção del (GJB6-D13S1830) no gene GJB6, identificada em 1% dos casos, sempre associada com a mutação 35delG no gene GJB223.
Em vários países da Europa, a prevalência de heterozigotos para 35delG tem sido estimada entre 2 a 4% da população com audição normal6,17. Foi encontrado 1 em 51(1,9%) caucasoides com a mutação 35delG, em estudo realizado no Brasil, similar à maioria das populações europeias24. Outro estudo, realizado com neonatos no Estado de São Paulo, mostrou que 35delG foi encontrada em 1 de 103(0,97%) neonatos25.
O estabelecimento das prevalências das mutações no país pode auxiliar na implantação de testes rápidos de diagnósticos moleculares que auxiliarão na conduta do médico em relação ao tratamento do paciente, acompanhado do aconselhamento genético às famílias dos membros afetados. Dessa forma torna-se fundamental o conhecimento da diversidade genética associada a esta afecção para se estabelecer melhores propostas de diagnóstico molecular para cada grupo populacional26.
Esse trabalho teve como objetivo estimar as prevalências das mutações 35delG e D13S1830 nos genes GJB2 e GJB6, respectivamente, em uma amostra formada por portadores de deficiência auditiva neurossensorial bilateral pré-lingual não-sindrômica de causa desconhecida, na população do Espírito Santo (sudeste do Brasil).
MATERIAIS E MÉTODOSPacientes e amostrasFoi realizado um estudo de corte transversal, no qual 77 portadores de deficiência auditiva, não aparentados, foram estudados, sendo 38 homens e 39 mulheres. A idade dos pacientes variou de 1 a 52 anos de idade. Todos residentes no estado do Espírito Santo.
Os pacientes provenientes de escolas orais auditivas e centros de apoio ao deficiente de várias regiões do Espírito Santo foram submetidos a uma anamnese para investigação da idade de início da deficiência auditiva, presença de outros casos na família, confirmação de surdez não-sindrômica e exclusão de causas ambientais: infecções pré-natais como rubéola, toxoplasmose, herpes, e infecções pós-natais, particularmente as meningites bacterianas e também a exposição a drogas ototóxicas. Após avaliação audiológica, exames de imitanciometria, audiometria tonal, e/ou audiometria comportamental e emissão otoacústica, constatou-se que todos os pacientes apresentavam surdez sensorial bilateral de moderada a profunda.
Foram coletados 3ml de sangue periférico com prévia obtenção do termo de consentimento livre e esclarecido dos pacientes ou responsáveis. O protocolo para este estudo foi aprovado pelo Comitê de Ética em Pesquisa (CEP) do Centro Integrado de Atenção a Saúde (CIAS)(Protocolo nº121/2006). O DNA genômico foi extraído utilizando-se kit de extração de DNA comercial (Puregene DNA Purification Kit - Gentra Systems), de acordo com instruções do fabricante.
Triagem das mutaçõesA amplificação da região genômica para análise da mutação 35delG no gene GJB2 foi realizada com um par de primers (F: 5 TCT TTT CCA GAG CAA ACC GC 3 e R: 5 GCT GGT GGA GTG TTT GTT CAC ACC CGC 3 ) e 65ºC de temperatura de anelamento. O produto de PCR apresentando 89 pb foi digerido com a enzima de restrição BstNI, caso não haja a deleção a enzima produz dois fragmentos (69pb e 20pb), se houver a deleção a enzima não corta o DNA (89pb). Os fragmentos produzidos após a digestão foram analisados através de eletroforese em gel de poliacrilamida 8%27.
A mutação del (GJB6-D13S1830) foi rastreada através da técnica PCR multiplex, utilizando três primers (F: 5 TTT AGG GCA TGA TTG GGG TGA TTT - 3; R1: 5 CAC CAT GCG TAG CCT TAA CCA TTTT - 3; R2: 5' TCA TCG GGG GTG TCA ACA AACA - 3') e 62ºC de temperatura de anelamento20,21. O primer F e o R1 foram utilizados para a detecção da presença da deleção e o primer R2 para detecção do alelo normal, que amplifica um fragmento dentro da região deletada. Quando os três primers são usados juntos, dois produtos de PCR podem ser obtidos, permitindo assim a distinção entre indivíduos homozigotos, heterozigotos, ou homozigotos normais. O produto amplificado foi analisado em eletroforese em gel de agarose 1,5%.
RESULTADOSNesse estudo, um total de 308 indivíduos com deficiência auditiva foi avaliado inicialmente, destes, 77 não apresentaram causas ambientais para o quadro clínico observado e foram incluídos no estudo (25% dos casos avaliados).
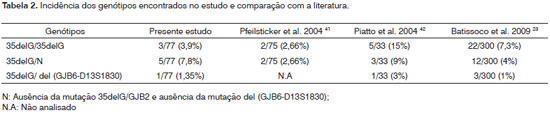
Os 77 indivíduos com surdez idiopática foram submetidos ao rastreamento molecular da mutação 35delG no gene GJB2 e da mutação del (D13S1830) no gene GJB6. A mutação 35delG foi encontrada em homozigose em 3 pacientes (3,9% dos casos analisados), sendo esta a etiologia da deficiência auditiva nestes indivíduos. Em 5 indivíduos a mutação 35delG foi encontrada em heterozigose (6,5% dos casos), porém a presença de somente um alelo mutante não justifica a causa da surdez nestes pacientes. Todos os pacientes normais e heterozigotos para a mutação 35delG também foram investigados para a mutação del (GJB6-D13S1830). Dos 74 indivíduos avaliados, um apresentou genótipo heterozigoto para a referida mutação (1,35% dos casos). O paciente heterozigoto para a mutação del (D13S1830/GJB6) também foi heterozigoto para a mutação 35delG/GJB2, concluindo a etiologia digênica da surdez no referido paciente. Em 68 pacientes não foi evidenciada a presença das referidas mutações, porém outras mutações, relacionadas ou não a Cx26 e Cx30, podem estar relacionadas com a causa do quadro clínico nestes pacientes. A Tabela 1 mostra os resultados do estudo.

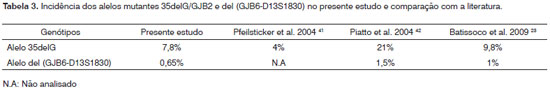
A frequência do alelo mutante 35delG/GJB2 na amostra foi 7,8%, e do alelo mutante del (D13S1830/GJB6) foi 0,65%.
DISCUSSÃOA heterogeneidade genética da deficiência auditiva não-sindrômica complica seu diagnóstico molecular, devido à grande quantidade de mutações já descritas em genes relacionados com a surdez e também devido à diversidade na predominância de cada alteração em populações diferentes.
A mutação 35delG no gene GJB2 é a principal causa da surdez genética em populações de origem caucasiana, podendo estar presente no estado homozigoto ou em heterozigose composta (com outras mutações no gene GJB2 ou no GBJ6)4,7. A mutação del (GJB6/D13S1830) é a segunda mutação mais frequente relacionada à DFNB em populações europeias, populações judaicas, e também no Brasil7,23.
A frequência do alelo mutante 35delG/GJB2 pode variar em diferentes regiões do mundo, como: Estados Unidos (1,0%); Austrália (1,0%); Áustria (1,7%); Turquia (1,8%); Portugal (2,2%); Espanha (2,5%), França (2,7%), Itália (2-4%)28,13,29,30,17,31.
No Brasil, provavelmente devido à grande miscigenação do país, a mutação 35delG/GJB2 não é rara também. Três estudos realizados no estado de São Paulo mostraram frequências de portadores do alelo mutante variando de 0,97% a 2,24%22,25,32.
Uma pesquisa realizada em 10 cidades de diferentes regiões do país revelou os seguintes dados quanto à frequência de 35delG na população brasileira: região norte 2,1%; região sudeste 1,5%; região sul 1,2% e região nordeste 0,8% portadores da mutação, embora as diferenças observadas não tenham sido significantes33.
Nesse estudo, a mutação 35delG/GJB2 e a mutação del (D13S1830/GJB6) foram estudadas em pacientes com surdez de causa idiopática residentes no estado do Espírito Santo/Brasil. Dos 77 indivíduos analisados, nove indivíduos não relacionados apresentaram a mutação 35delG/GJB2 e, destes, um apresentou a mutação em heterozigose composta com a mutação D13S1830 no gene GJB6. A frequência dos alelos mutantes 35delG/GJB2 e del (GJB6-D13S1830) na amostra foram 7,8% e 0,65%, respectivamente. Nossos dados estão em concordância com estudos prévios realizados na população brasileira (Tabela 2 e 3) e também com muitos outros estudos os quais documentam a incidência de mutações nos genes GJB2 e GJB6 em pacientes com surdez pré-lingual não-sindrômica.28,29,34-40.
Embora a análise molecular da deficiência auditiva ainda seja muito escassa nos países em desenvolvimento, a pesquisa de mutações nos genes GJB2 e GJB6 é de fundamental importância para a saúde pública e aconselhamento genético. A proporção de deficientes auditivos devido a causas genéticas tende a aumentar como resultado do aumento de investimentos e melhorias nos sistemas de saúde dos países em desenvolvimento como o Brasil. O estabelecimento da prevalência e tipos de mutações que causam a perda auditiva não-sindrômica no Brasil, como no presente estudo, pode auxiliar na implantação de modelos simples e específicos para detecção das principais mutações responsáveis pela surdez genética no país. O diagnóstico molecular propicia um aconselhamento genético preciso aos membros das famílias e permite uma reabilitação precoce nas crianças dessas famílias.
CONCLUSÃOOs dados obtidos no presente estudo confirmaram a existência da mutação 35delG no gene GJB2 em casos de perda auditiva neurossensorial bilateral não-sindrômica de moderada a profunda na população do Espírito Santo/Brasil, resultado que concorda com a literatura. A mutação del (GJB6-D13S1830) foi encontrada em heterozigose composta com a mutação 35delG/GJB2 em um paciente. Esses achados reforçam a importância do diagnóstico genético que, além de elucidar a etiologia da perda auditiva do paciente, também pode propiciar um tratamento precoce para crianças e aconselhamento genético para a família dos afetados.
REFERÊNCIAS BIBLIOGRÁFICAS1. Downs MP. Universal newborn hearing screening - the Colorado story. Int J Pediatr Otorhinolaryngol. 1995;32:257-9.
2. Mehl AL, Thomson V. Newborn hearing screening: the great omission. Pediatrics. 1998;101:E4.23.
3. Mehl AL, Thomson V. The Colorado newborn hearing screening project, 1992-1999: on the threshold of effective population- based universal newborn hearing screening. Pediatrics. 2002;109:E7.
4. Piatto VB, Maniglia JV. Importância do Gene Conexina 26 na etiologia da deficiência auditiva sensorioneural não-sindrômica. Acta Awho. 2001;20(2):106-12.
5. Bitner-Glindzicz M. Hereditary deafness and phenotyping in humans. Br Med Bull. 2002;63:73-94.
6. Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, DAgruma L, et al. Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet.1998;351:394-8.
7. Gabriel H, Kupsch P, Sudendey J, Winterhager E, Jahnke K, Lautermann J. Mutations in the connexin26/GJB2 gene are the most common event in non-syndromic hearing loss among the German population. Hum Mutat. 2001;17:521-2.
8. Lench N, Houseman M, Newton V, Van Camp G, Mueller R: Connexin- 26 mutations in sporadic non-syndromal sensorineural deafness. Lancet. 1998;351:415.
9. Morell RJ, Kim HJ, Hood LJ, Goforth L, Friderici K, Fisher R, et al.: Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med. 1998;339:1500-05.
10. Ohtsuka A, Yuge I, Kimura S, Namba A, Abe S, Van Laer L, et al. GJB2 deafness gene shows a specific spectrum of mutations in Japan, including a frequent founder mutation. Hum Genet. 2003;112:329-33.
11. Park HJ, Hahn SH, Chun YM, Park K, Kim HN. Connexin26 mutations associated with nonsyndromic hearing loss. Laryngoscope. 2000;110:1535-8.
12. Rabionet R, Zelante L, Lopez-Bigas N, DAgruma L, Melchionda S, Restagno G, et al. Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum Genet. 2000;106:40-4.
13. Wilcox SA, Saunders K, Osborn AH, Arnold A, Wunderlich J, Kelly T, et al. High frequency hearing loss correlated with mutations in the GJB2 gene. Hum Genet. 2000;106:399-405.
14. Goodenough DA, Goliger JA, Paul DL. Connexins, connexons, and intercellular communication. Annu Rev Biochem. 1996;65:475-502.
15. Kumar NM, Gilula NB. The gap junction communication channel. Cell 1996;84:381-8.
16. Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1-27.
17. Gasparini P, Rabionet R, Barbujani G, Melchionda S, Petersen M, Brondum-Nielsen K, et al. High carrier frequency of the 35delG deafness mutation in European populations. Eur J Hum Genet. 2000;8:19-23.
18. Rabionet R, Gasparini P, Estivill X. Molecular genetics of hearing impairment due to mutations in gap junction genes encoding beta connexins. Hum Mutat. 2000;16(3):190-202. Review.
19. Kenneson A, Van Naarden Braun K, Boyle C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review. Genet Med. 2002;4:258-74.
20. Del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Tellería D, et al. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med. 2002;346(4):243-9.
21. Del Castillo I, Moreno-Pelayo MA, Del Castillo FJ, Brownstein Z, Marlin S, Adina Q, et al. Prevalence and evolutionary origins of the del(GJB6-D13S1830) mutation in the DFNB1 locus in hearing- impaired subjects: a multicenter study. Am J Hum Genet. 2003;73(6):1452-1458. Epub 2003 Oct 21.
22. Piatto VB, Bertollo EM, Sartorato EL, Maniglia JV. Prevalence of the GJB2 mutations and the del(GJB6-D13S1830) mutation in Brazilian patients with deafness. Hear Res. 2004;196:87-93.
23. Batissoco AC, Abreu-Silva RS, Braga MC, Lezirovitz K, Della-Rosa V, Alfredo T Jr, et al. Prevalence of GJB2 (connexin-26) and GJB6 (connexin-30) mutations in a cohort of 300 Brazilian hearing-impaired individuals: implications for diagnosis and genetic counseling. Ear Hear. 2009;30(1):1-7
24. Gasparini P, Estivill X, Volpini V, Totaro A, Castellvi-Bel S, Govea N, et al. Linkage of DFNB1 to non-syndromic neurosensory autosomal-recessive deafness in Mediterranean families. Eur J Hum Genet. 1997;5:83-8.
25. Sartorato EL, Gottardi E, de Oliveira CA, Magna LA, Annichino-Bizzacchi JM, Seixas CA, Maciel-Guerra AT, et al. Determination of the frequency of 35delG allele in Brazilian neonates. Clin Genet. 2000;58(1):339-40.
26. Smith RJ, Hone S. Genetic screening for deafness. Pediatr Clin North Am. 2003;50:315-29.
27. Wilcox SA, Osborn AH, Dahl HH. A simple PCR test to detect the common 35delG mutation in the connexin 26 gene. Mol Diagn. 2000;5(1):75-8.
28. Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, et al. Novel mutations in the connexin 26 gene (GJB2) that cause autossomal recessive (DFNB1) hearing loss. Am J Hum Genet. 1998;62:792-9.
29. Frei K, Szuhai K, Lucas T, Weipoltshammer K, Schöfer C, Ramsebner R, et al. Connexin 26 mutations in cases of sensorineural deafness in eastern Áustria. Eur J Hum Genet. 2002;10:427-32.
30. Tekin M, Akar N, Cin S, Blanton SH, Xia XJ, Liu XZ, et al. Connexin 26 (GJB2) mutations in the Turkish population: implications for the origin and high frequency of the 35delG mutation in Caucasians. Hum Genet. 2001;108:385-9.
31. Lucotte G, Bathelier C, Champenois T. PCR test for diagnosis of the common GJB2 (connexin 26) 35delG mutation on dried blood spots and determination of the carrier frequency in France. Mol Cell Probes. 2001;15:57-9.
32. Oliveira CA, Alexandrino F, Abe-Sandes K, Silva WA Jr, Maciel-Guerra AT, Magna LA, et al. Frequency of the 35delG mutation in the GJB2 gene in samples of European, Asian and African Brazilians. Hum Biol. 2004;76(2):313-16.
33. Oliveira CA, Pimpinati CJ, Alexandrino F, Magna LA, Maciel-Guerra AT, Sartorato EL. Allelic frequencies of the 35delG mutation of the GJB2 gene in different Brazilian regions. Genet Test. 2007;11:1-3.
34. Green GE, Scott DA, McDonald JM, Woodworth GG, Sheffield VC, Smith RJ. Carrier rates in the mid-western United States for GJB2 mutations causing inherited deafness. J Am Med Ass. 1999;281:2211-16.
35. Sobe T, Vreugde S, Shahin H, Berlin M, Davis N, Kanaan M, et al. The prevalence and expression of inherited connexin 26 mutations associated with non-syndromic hearing loss in the Israeli population. Hum Genet. 2000;106:50-7.
36. Mustapha M, Salem N, Delague V, Chouery E, Ghassibeh M, Rai M, et al. Autosomal recessive non-syndromic hearing loss in theLibanese population: prevalence of the 30delG mutation and report of two novel mutations in the connexin 26 (GJB2) gene. J Med Genet. 2001;38(10):36.
37. Liu XZ, Xia XJ, Ke XM, Ouyang XM, Du LL, Liu YH, et al. The prevalence of connexin 26 (GJB2) mutations in the Chinese population. Hum Genet. 2002;111:394-7.
38. Najmabadi H, Cucci RA, Sahebjam S, Kouchakian N, Farhadi M, Kahrizi K, et al. GJB2 mutations in Iranians with autosomal recessive nonsyndromic sensorineural hearing loss. Hum Mutat. 2002;19:572-7.
39. Pampanos A, Economides J, Iliadou V, Neou P, Leotsakos P, Voyiatzis N, et al. Prevalence of GJB2 mutations in prelingual deafness in the Greek population. Int J Pediatr Otorhinolaryngol. 2002;65:101-8.
40. Shahin H, Walsh T, Sobe T, Lynch E, King MC, Avraham KB, et al. Genetics of congenital deafness in the Palestinian population:multiple connexin 26 alleles with shared origins in the Middle East. Hum Genet. 2002;110:284-9.
41. Pfeilsticker LN, Stole G, Sartorato EL, Delfino D, Guerra ATM. A investigação genética na surdez hereditária não-sindrômica. Rev Bras Otorrinolaringol. 2004;70(2):181-6.
42. Piatto VB, Bertollo EMG, Sartorato EL, Maniglia JC. Prevalence of the GJB2 mutations and the del (GJB6-D13S1830) mutations in Brazilian patients with deafness. Hear Res. 2004;196(1-2):87-93.
1. Mestre, Professora das Faculdades Integradas São Pedro, AEV/FAESA e Pós-graduanda, em nível de Doutorado, no Programa de Pós-graduação em Biotecnologia - RENORBIO/UFES.
2. Graduação em Ciências biológicas, Pesquisadora.
3. Graduação em Ciências biológicas, Pesquisadora.
4. Graduação em Ciências biológicas, Pesquisadora.
5. Doutora, Coordenadora de Pesquisa e Extensão das Faculdades Integradas São Pedro, AEV/FAESA.
Faculdades Integradas São Pedro (AEV/FAESA)
Endereço para correspondência:
Melissa de Freitas Cordeiro-Silva
Rua Maria Eleonora Pereira 1171/301
Vitória ES 29060-180
Tel. (0xx27) 2122-4597
E-mail: melissasilva@aev.edu.br
Fundação de Apoio à Ciência e Tecnologia do Espírito Santo - FAPES
Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) da BJORL em 10 de agosto de 2009. cod 6558
Artigo aceito em 28 de outubro de 2009
