Introdução Em 1931, Klinger1 descreveu um caso de um paciente com doença de vias aéreas superiores, morto após insuficiência renal. Em 1936, Friedrich Wegener2 relatou um caso idêntico e, em 1939, reportou a primeira série de casos da moléstia que ficaria conhecida como Granulomatose de Wegener (GW). Fiemberg3, em 1953, introduziu conjuntamente com Carrington e Liebow4 o conceito da Granulomatose de Wegener limitada. Eles observaram que a doença poderia restringir-se a apenas um ou mais órgãos e, não necessariamente, apresentaria um envolvimento das três regiões-alvo (vias aéreas superiores, inferiores e rim). Este conceito foi reforçado subseqüentemente por Cassan e seus colaboradores5.
Em 1954, Godman e Churg6 delinearam os três critérios clássicos para o diagnóstico da granulomatose de Wegener: a presença de lesões granulomatosas necrotizantes no trato respiratório, vasculite e glomerulonefrite.
Granulomatose de Wegener é uma doença sistêmica idiopática rara, imunologicamente mediada, caracterizada por acometer as pequenas artérias dos tratos respiratórios superior e inferior e do rim, provocando reação inflamatória com necrose, formação de granuloma e vasculite nestes órgãos. Estudos americanos demonstram uma prevalência de 3 casos para cada 100.000 pessoas7,8. A idade média do diagnóstico costuma estar entre 20 e 40 anos e o sexo masculino é mais acometido do que o feminino numa proporção de 1,5:1,0. É uma doença incomum em pessoas da raça negra9.
O diagnóstico baseia-se no quadro clínico, no exame anátomo-patológico dos órgãos envolvidos e na positividade do ANCA-c (antinuclear cytoplasmic antibodies). O tratamento é feito com drogas imunossupressoras e deve ser instituído o mais precocemente possível, pois trata-se de um enfermidade grave, que leva à morte devido a infecções e, principalmente, à falência renal.
Como sintomas gerais iniciais, o paciente costuma apresentar febre, anorexia, emagrecimento, fadiga e fraqueza9.
O trato respiratório alto está envolvido em 70 a 100% dos casos. As manifestações mais freqüentes envolvem o nariz e os seios paranasais (em 80 a 90% dos casos) e são: obstrução nasal crônica com rinorréia clara, ulceração e edema da mucosa nasal, parosmia, epistaxe e cefaléia. Perfuração do septo nasal e ulceração e erosão do vômer são clássicas. A deformidade do nariz "em sela", devido à destruição da cartilagem nasal e do vômer, ocorre em apenas 3,5 a 7% dos casos. Estas lesões são freqüentemente infectadas secundariamente por Staphylococcus aureus7,10,11.
Podem ocorrer otite média secretora, otite média crônica, com perfuração da membrana timpânica, otalgia e otorréia. Ainda existem casos descritos de disacusia sensorioneural, vertigens e tinnitus em pacientes com granulomatose de Wegener12..
A prevalência do envolvimento otológico está em torno de 35%, e relaciona-se à vasculite e ao bloqueio da tuba auditiva por acometimento da rinofaringe7,12.
Paralisia facial é encontrada em 8 a 10% dos casos e resulta de vasculite do vasa - vasorum do nervo facial, ou de invasão granulomatosa do tecido da orelha média, ou de lesão granulomatosa primária do nervo ou da combinação destes fatores.
O envolvimento pulmonar é também extremamente comum (70 a 90% dos casos). Tosse produtiva, dispnéia, hemoptise, dor e desconforto torácico são os principais sintomas. Anormalidades nas radiografias do tórax são vistas em mais de 90% dos casos, e incluem lesões nodulares escavadas não calcificadas, largas, múltiplas e bilaterais.
As características nefrológicas da granulomatose de Wegener são predominantemente representadas por glomerulonefrite focal necrotizante, que leva à falência renal e morte. Anormalidades renais são encontradas em 50 a 90% dos casos. Hematúria, leucocitúria e anormalidades nas mensurações de uréia e creatinina são as alterações encontradas nos exames dos doentes.
Manifestações cutâneas (petéquias e lesões ulceradas) ocorrem em aproximadamente 40% dos pacientes, nas extremidades dos membros inferiores.
Mialgias e artralgias migratórias são freqüentes (40 a 70%), mas efêmeras.
Acometimento ocular, cardíaco e do sistema nervoso central ocorrem em cerca de 40% dos casos e são inespecíficos.
Disfonia, estridor laríngeo, sibilos, ulceração e edema oral e gengivite complementam o acometimento de cabeça e pescoço7.
O padrão-ouro para o diagnóstico da granulomatose de Wegener é a descoberta de vasculite granulomatosa necrotizante no exame anátomo-patológico do tecido doente biopsiado. A necrose costuma ser extensa, com "debris" leucocíticos e microabscessos polimorfonucleares. As lesões granulomatosas apresentam polimorfonucleares, células plasmáticas, raros eosinófilos e linfócitos e, especialmente, células gigantes multinucleadas. A vasculite é caracterizada pelo envolvimento de arteríolas e vênulas, com conseqüente oclusão da luz vascular10.
O tecido pulmonar, obtido preferencialmente por toracotomia aberta, oferece o mais alto rendimento diagnóstico, revelando quase sempre uma vasculite granulomatosa necrotizante10,11.
O achado de títulos elevados de ANCA-c serve para consubstanciar o diagnóstico. Os ANCA-c são anticorpos circulantes contra elementos do núcleo do citoplasma de neutrófilos e monócitos. A imunofluorescência indireta é o melhor método para titulação do ANCA-c. Valores a partir de 1/16 são considerados positivos7,9-11.
Muitos investigadores acreditam que o ANCA-c é altamente específico para granulomatose de Wegener, com 98% de especificidade e com sensibilidade variando entre 34 e 92%. Entretanto, outros autores relatam positividade do ANCA-c em outras vasculites e síndromes autoimunes, como a poliarterite nodosa e a doença de Kawasaki7.
A granulomatose de Wegener limitada apresenta menor positividade do ANCA-c (60% dos casos ), assim como GW em remissão9.
O diagnóstico baseia-se na história clínica, nos achados de exame físico, radiológicos, tomográficos, anátomo-patológicos e na positividade do ANCA-c9,4.
Diagnósticos positivos são raramente feitos em formas muito precoces de GW e em casos de GW limitada9.
Em sua manifestação típica, o complexo clínico-patológico clássico da GW permite habitualmente a diferenciação imediata de outros distúrbios, tais como poliarterite nodosa, síndrome de Churg Strauss, linfoma, abuso de cocaína, granuloma médio-facial, síndrome de Goodpasture, tuberculose e demais doenças granulomatosas9-12.
O tratamento da GW está baseado na utilização de drogas imunossupressoras. Preconiza-se o uso de ciclofosfamida, administrada nas doses de 2 mg/ Kg/ dia, por via oral. A contagem de Leucócitos deve ser monitorizada atentamente durante a terapia, e a posologia ajustada de forma a manter a contagem de neutrófilios acima de 3.000/mm³. A ciclofosfamida deve ser continuada por 1 ano após a indução da remissão completa e, daí em diante, será reduzida gradualmente até poder ser suspensa.
No início da terapia, os glicocorticóides devem ser administrados juntamente com a ciclofosfamida. Em geral, é utilizada a prednisona, 1 mg/ Kg/ dia, inicialmente (no 1º mês de terapia), segundo um esquema diário, com conversão gradual para um programa de dias alternados, seguido por redução e suspensão após aproximadamente 6 meses.
Alguns relatos indicam um efeito benéfico da associação sulfametoxazol-trimetoprim no controle das infecções secundárias da GW, porém não existem dados concretos para confirmá-lo, particularmente nos pacientes com doença renal e pulmonar graves.
A sobrevida média dos doentes é de 8 anos e meio; a sobrevida em 10 anos é estimada em 40%, se houver envolvimento do rim e, em 60 a 70% se não houver comprometimento renal7.
Apresentação de caso clínico
E.I.P., com 18 anos, do sexo feminino, branca. Há 2 meses, iniciou quadro de cefaléia frontal e em hemiface esquerda, em peso, sem irradiação, de início insidioso, com piora progressiva, acompanhada de obstrução nasal, rinorréia purulenta anterior e posterior, epistaxe, cacosmia, e febre não medida. Procurou atendimento médico com 5 dias de história, quando foi tratada com amoxicilina por 10 dias, devido à suspeita de rinossinusite aguda. Evoluiu com piora do estado geral, febre alta, dor torácica, dispnéia, tosse produtiva com expectoração esverdeada, otalgia, otorréia e hipoacusia bilateral. Nesta ocasião, procurou novamente auxílio médico, e foi então internada para melhor avaliação e tratamento no Hospital Municipal Universitário de São Bernardo do Campo.
Apresentava-se com emagrecimento de 8 Kg, astenia, vertigem e artralgia em joelho direito. Não havia nenhum antecedente pessoal ou familiar relevante.
Ao exame físico geral apresentava-se descorada ++/4, desidratada +/4, afebril e taquicárdica (FC: 120 bpm). Ao exame otorrinolaringológico, apresentava perfuração central ampla de membrana timpânica bilateralmente, com otorréia em grande quantidade, deformidade do dorso nasal "em sela", abundantes crostas acastanhadas em ambas as fossas nasais, septo nasal íntegro mas com mucosa de aspecto granuloso, conchas nasais inferiores e médias hipotróficas. Boca e orofaringe sem alterações.
À ausculta pulmonar: murmúrio vesicular presente bilateralmente, com sibilos esparsos discretos, roncos e estertores creptantes difusos, principalmente em hemitórax direito.
Havia petéquias em membros inferiores. Demais aparelhos sem alterações.
Foi solicitada uma série de exames subsidiários para tentar se estabelecer o diagnóstico, a etiologia e a extensão da doença. O hemograma mostrava uma anemia normocrômica normocítica e leucocitose sem desvio à esquerda (19.500 leucócitos).
A radiografia e a tomografia computadorizada de tórax apresentavam uma imagem sugestiva de abscesso em terço médio e superior de pulmão direito; calcificação junto ao hilo pulmonar direito, focos pneumônicos à esquerda e derrame pleural bilateral.
Na tomografia computadorizada de seios paranasais, notava-se um espessamento do revestimento mucoso da fossa nasal; comunicação dos seios maxilares com a cavidade nasal bilateralmente, conchas nasais inferiores e médias hipotróficas; material com densidade de partes moles nos seios maxilares, etmoidais, esfenoidais e frontais (Figuras 1, 2, 3 e 4 ).
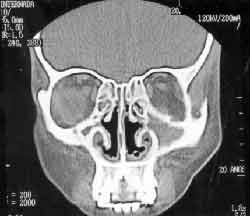
Figura 1
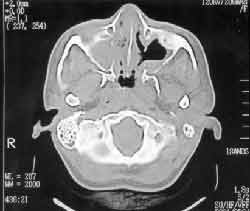
Figura 2
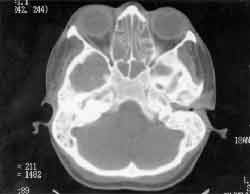
Figura 3
Figura 4
A pesquisa de BAAR no escarro resultou negativa em várias amostras. O exame de PPD, a sorologia para o HIV e o FTA-ABS também resultaram negativos. A cultura do escarro foi positiva para Pseudomonas e negativa para fungos. A cultura do líquido pleural mostrou-se negativa para BAAR e fungos.
À broncoscopia visualizou-se lesão ulcerada e compressão extrínseca do brônquio fonte direito. Foi então realizada biópsia da lesão, cujo exame anátomo patológico (A-P) demonstrou extensas áreas de necrose e exsudato de neutrófilos.
O exame A-P de biópsia realizada em mucosa nasal resultou em processo inflamatório crônico inespecífico.
A titulação do ANCA-c teve resultado positivo (1/32).
Após a positividade do ANCA-C, optou-se por nova biópsia de mucosa nasal, mas, desta vez, sob visão microscópica, para se firmar o diagnóstico de granulomatose de Wegener. O A-P revelou fragmentos de mucosa respiratória parcialmente revestidos por epitélio pavimentoso estratificado, apresentando no córion, numerosas estruturas vasculares de pequeno calibre, cuja parede estava permeada por neutrófilos e eosinófilos, entremeados por edema e infiltrado misto. Alguns vasos com nítida agressão da parede, com áreas de necrose fibrinóide. O quadro histológico sugeria vasculite granulomatosa necrotizante.
A função renal, pesquisada pela dosagem de uréia e creatinina, estava preservada. O exame de urina I mostrava uma micro-hematúria discreta.
À audiometria tonal, detectou-se uma disacusia condutiva leve a severa bilateral, ascendente e simétrica. Havia boa discriminação à audiometria vocal.
A paciente evoluiu com piora clínica, radiográfica e tomográfica, mesmo com antibioticoterapia de amplo espectro. Vários antimicrobianos bactericidas e bacteriostáticos foram introduzidos, levando-se em conta o quadro clínico e os resultados dos exames subsidiários realizados pela doente. Ceftriaxone, clindamicina, vancomicina, metronidazol, imipenem, amicacina, tazocin, anfotericina B e esquema tríplice para tuberculose foram utilizados seqüencialmente pela clínica médica para tentar conter o quadro infeccioso, sem sucesso. Após a positividade do ANCA-c e o resultado da segunda biópsia de mucosa nasal, firmou-se o diagnóstico de granulomatose de Wegener e institui-se o tratamento com ciclofosfamida e prednisona. No início deste tratamento, a paciente foi submetida a pneumectomia parcial à direita, por extenso acometimento e destruição de seu pulmão direito. Após a cirurgia e a continuidade da terapia imunossupressora, a paciente apresentou melhora importante e obteve alta hospitalar depois de 15 dias.
Discussão A paciente não se enquadrava no perfil mais comum para que houvesse a suspeita de granulomatose de Wegener. Embora com quadro clínico suspeito, o fato de ser do sexo feminino e adolescente contrariava os dados da literatura.
Em um primeiro momento, levantou-se a hipótese de que se tratasse de uma tuberculose associada a infecção bacteriana secundária em vias aéreas superiores e inferiores, o que não foi confirmado através de exames subsidiários. A introdução do esquema tríplice para tratamento de tuberculose também não alterou o curso da doença.
As biópsias do tecido nasal e brônquico inconclusivas afastaram, a princípio, a possibilidade de granulomatose de Wegener. O fato das vias urinárias não estarem acometidas também deixou este diagnóstico menos provável.
No entanto, após a positividade do ANCA-c, optou-se por nova biópsia nasal, desta vez guiada por visão microscópica, para extração de material da mucosa nitidamente doente. Com o achado de vasculite granulomatosa necrotizante, típico de granulomatose de Wegener, fechou-se o diagnóstico e instituiu-se o tratamento imunossupressor, com observação de melhora importante do quadro.
Conclusões Granulomatose de Wegener é uma entidade rara e fazer o seu diagnóstico torna-se um verdadeiro desafio para o médico. A hipótese de sua ocorrência deverá ser sempre lembrada quando houver sintomas relacionados com acometimento das vias aéreas superiores, inferiores e do rim. Nossa paciente, no entanto, não apresentava comprometimento renal, enquadrando-se na granulomatose de Wegener limitada, o que tornou o diagnóstico ainda mais improvável.
O quadro da doente teve evolução dramática e arrastada até proceder-se o diagnóstico, fazendo inclusive com que esta tivesse parte de seu pulmão direito extraída, o que poderia ter sido evitado se houvesse uma conclusão anterior da causa da moléstia.
Sugerimos que as biópsias nasais sejam feitas sob visão microscópica ou endoscópica, evitando falhas diagnósticas por material insuficiente ou saudável, o que atrasa substancialmente a definição da doença, aumentando as seqüelas do processo granulomatoso.
É de suma importância que o diagnóstico seja firmado o mais precocemente possível, para que a instituição do tratamento imunossupressor seja breve e a remissão se torne possível, reduzindo-se assim a morbi-mortalidade da doença.
Referências Bibliográficas 1. Klinger H. Grenzformen der Periarteritis nodosa. Frankf Z Pathol 1931;42:455-480.
2. Wegener F. Uber generalisierte, septische Gefässerkrankungen. Verh Dtsch Ges Pathol 1936;29:202-210.
3. Fienberg R. Necrotizing granulomatosis and angiitis of the lungs. Am J Clin Pathol 1953;23:413-428.
4. Carrington CB, Liebow M. Limited forms of angiitis and granulomatosis of Wegener's type. Am J Med 1966;41:497-527.
5. Cassan SM, Coles DT, Harisson EG. The concept of limited forms of Wegener's granulomatosis. Am J Med 1970;49:366-379.
6. Godman GC, Churg J. Wegener's granulomatosis. Pathology and review of the literature. Arch Pathol 1954;58:533-553.
7. Hartl DM, Aidan P, Brugière O, Sterkers O. Wegener's Granulomatosis Presenting as a Recurrence of Chronic Otitis Media. Am J Otol 1998;19(1):54-60.
8. Koldingsnes W, Nossent H. Epidemiology of Wegener's Granulomatosis in Nothern Norway. Art Rheum 2000;43 (11):2481-2487.
9. Devaney KO, Ferlito A, Devaney SL, Hunter BC, Rinaldo A. ClinicoPathological Consultation: Wegener's Granulomatosis of the Head and Neck. Ann Otol Rhinol Laryngol 1998;107:439-445.
10. Jennings CR, Jones NS, Dugar J, Powell RJ, Lowe J. Wegener's Granulomatosis - A review of diagnosis and treatment in 53 subjects. Rhinology 1998;36:188-191.
11. Jones NS. Nasal manifestations of rheumatic diseases. Ann Rheum Dis 1999;58:589-590.
12. Scully RE, Mark EJ, Mcneely WF, Ebeling SH. Case Records of the Massachussets general Hospital. N Engl J Med 1999;340(17): 1349-1353.
[1] Residentes do terceiro ano de Otorrinolaringologia da Faculdade de Medicina da FUABC.
[2] Residente do primeiro ano de Otorrinolaringologia da Faculdade de Medicina da FUABC.
[3] Auxiliar de ensino da Disciplina de Otorrinolaringologia da Faculdade de Medicina da FUABC.
[4] Professora Titular da Disciplina de Otorrinolaringologia da Faculdade de Medicina da FUABC.
Disciplina de Otorrinolaringologia da Faculdade de Medicina da FUABC - Av. Príncipe de Gales 821 Santo André SP Brasil 09060-650 - Tel (0xx11)449-3347
Endereço para correspondência: Rubens E. C. Rodrigues - Rua Estrada Velha da Penha 88 apto 72 bloco 2, 03090-020 Tatuapé São Paulo SP.
