INTRODUÇÃOA deficiência auditiva (DA) é o déficit sensorial mais comum, e resulta na restrição das habilidades de se comunicar pela linguagem falada1, trazendo ainda problemas sociais, psíquicos e educacionais a seus portadores. Quando o diagnóstico é realizado no primeiro ano de vida, possibilita intervenção médica e/ou fonoaudiológica ainda no período crítico de maturação e plasticidade funcional do sistema nervoso central, com prognóstico mais favorável em relação ao desenvolvimento global da criança2.
Para o diagnóstico audiológico testes comportamentais e eletrofisiológicos estão disponíveis e podem ser utilizados como procedimentos de avaliação3-11. A bateria de testes a ser aplicada é determinada de acordo com a maturação de cada individuo.
A DA neurossensorial congênita pode ser de origem genética ou não-genética. Dentre os casos de origem genética há aqueles em que a DA faz parte de quadros sindrômicos (30% dos casos) e aqueles em que é um sinal isolado - a DA neurossensorial não-sindrômica (70%)12-14. Mais de 100 genes estão potencialmente envolvidos na DA pré-lingual não-sindrômica14, que em cerca de 85% dos casos é de herança autossômica recessiva, em 12 a 14% autossômica dominante e em 1 a 3% ligada ao cromossomo X15,16.
Dentre os casos de DA não-sindrômica de herança autossômica recessiva destacam-se aqueles decorrentes de mutações no gene GJB2, que codifica a proteína conexina 26. Uma dessas mutações, 35delG responde por cerca de 70% dos alelos mutantes em caucasóides europeus e norte-americanos, com uma freqüência de heterozigotos de 2,3% a 4%17-20. No Brasil, a freqüência de heterozigotos da mutação 35delG é estimada em 1:7421. Outras alterações genéticas que podem ser encontradas em portadores de DA não-sindrômica são deleções no gene da conexina 30 (GJB6-Cx30), del (GJB6 - D13S1830) e del (GJB6 - D13S1854), e também a mutação A1555G no gene mitocondrial 12SrRNA22-24.
Em países em desenvolvimento, como o Brasil, fatores ambientais como as infecções congênitas e intercorrências perinatais ainda são bastante comuns25. No entanto, com a melhoria da atenção à saúde materno-infantil a freqüência relativa dos casos de origem genética deve aumentar progressivamente26.
Nos casos de deficiência auditiva de origem genética, além da estimulação auditiva e da indicação do Aparelho de Amplificação Sonora Individual (AASI) o mais precocemente possível, o aconselhamento genético também deve ser parte da conduta, permitindo aos indivíduos ou famílias a tomada de decisões conscientes e equilibradas a respeito da procriação27. A terapia gênica está ainda em fase de pesquisa, e mesmo assim não se aplica a todas as situações28.
O aconselhamento genético em casos de DA hereditária não-sindrômica depende de um diagnóstico preciso a respeito do mecanismo de herança. A introdução, em nosso país, da pesquisa da mutação 35delG do gene GJB2 entre os exames subsidiários necessários ao esclarecimento do diagnóstico etiológico tem sido fundamental para que os objetivos do aconselhamento genético sejam cumpridos. De fato, sua detecção tem alta relevância no rastreamento das causas da DA, demonstrando que é fundamental que o estudo molecular seja feito de forma abrangente, a fim de evitar que diagnósticos precipitados prejudiquem a orientação da família.
O objetivo deste trabalho é relatar o perfil audiológico e os resultados da avaliação genética de uma família do município de São Paulo, salientando ainda a heterogeneidade genética nos casos de DA, assim como a complexidade de seu aconselhamento genético.
MATERIAL E MÉTODOEste estudo foi aprovado pelo Comitê de Ética em Pesquisa do CEFAC - Centro de Especialização em Fonoaudiologia Clínica, sob número 172/05.
O caso-índice (II.5, Figura 1), do sexo masculino, portador de DA foi atendido pela primeira vez aos 16 anos no módulo prático de Audiologia Clínica do Instituto de Estudos Avançados da Audição (IEAA) em novembro de 2004 para atualização de exame audiológico, a pedido da fonoaudióloga que já o atendia em terapia. Tratava-se do segundo dos quatro filhos de casal não-consangüíneo, mãe (I.2) com 41 anos e pai (I.3) com 40 anos, sendo uma irmã mais velha (II.4) de 17 anos com sensibilidade auditiva dentro dos padrões de normalidade, e que tinha um filho (III.2) de um ano de idade, e dois irmãos mais novos, um com cinco anos (II.6) e outro de três anos (II.7), também deficientes auditivos. Havia ainda uma meio-irmã (II.2) mais velha pelo lado materno de 21 anos, também com sensibilidade auditiva adequada, cuja filha (III.1) tinha um ano de idade. A DA dos três irmãos afetados foi de instalação pré-lingual e não progressiva.
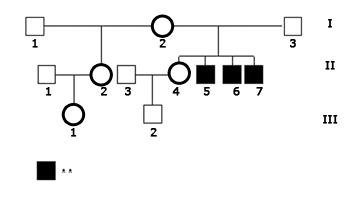
Figura 1. Heredograma da família estudada no presente trabalho. - Legenda: ** Perda Auditiva.
A avaliação audiológica inclui tanto os testes comportamentais quanto os eletrofisiológicos, realizados de acordo com a necessidade de cada participante, com relação à idade e a presença ou não de deficiência auditiva.
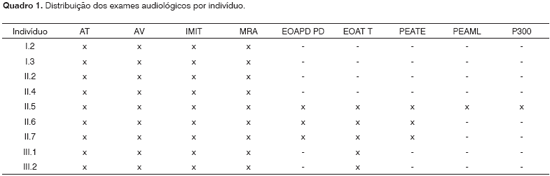
Foram realizados no IEAA audiometria tonal (AT) e audiometria vocal (AV), imitanciometria (IMIT), medida do reflexo acústico (MRA) e testes de Emissões Otoacústicas por Produto de Distorção (EOA PD) e Transiente (EOA T).
Os exames de Potencial Evocado Auditivo de Tronco Encefálico (PEATE), Potencial Evocado Auditivo de Média Latência (PEAML) e P300foram realizados no Centro de Docência e Pesquisa da Faculdade de Medicina da Universidade de São Paulo - CDP FMUSP. O Quadro 1 mostra os exames audiológicos realizados em cada membro da família.
Estudo molecularA avaliação genética foi realizada no Centro de Biologia Molecular e Engenharia Genética (CBMEG) da UNICAMP. A presença da mutação 35delG no gene GJB2 foi investigada por meio da técnica *AS-PCR (reação em cadeia da polimerase alelo-específico), que discrimina o alelo normal do mutante e permite distinguir homozigotos normais, homozigotos 35delG e heterozigotos portadores da mutação; foi realizado, ainda, seqüenciamento completo desse gene.
Também foram pesquisadas deleções envolvendo o gene da conexina 30 (GJB6-Cx30), del (GJB6 - D13S1830) e del (GJB6 - D13S1854) utilizando a técnica de PCR-multiplex, além da mutação A1555G no gene mitocondrial 12SrRNApor análise de restrição por meio de digestão com a enzima Bsm AI.
RESULTADOSA avaliação audiológica do caso-índice (II.5) e de seus irmãos (II.6 e II.7) revelou curva audiométrica com perda auditiva do tipo neurossensorial, bilateral, simétrica, e configuração descendente acentuada. Nos indivíduos II.5 e II.7 o grau de DA era moderadamente severo bilateralmente, e em II.6 moderado à direita e moderadamente severo à esquerda. O estudo audiológico dos pais (I.2 e I.3), irmã (II.4), meio-irmã (II.2) e sobrinhos (III.1 e III.2) dos três pacientes revelou média dos limiares auditivos dentro dos padrões de normalidade. A EOAPD estava ausente em II.6 e a EOAT estava ausente no indivíduo II.7.
O exame de PEATE foi compatível com perda auditiva severa para clicks bilateralmente em II.5 e II.7 e moderadamente severa em II.6. Em II.5 o P300 mostrou latências dentro da normalidade bilateralmente.
Quanto ao estudo molecular, nenhum dos indivíduos analisados apresentou deleções no gene da conexina 30, nem mutação no gene 12SrRNA. O estudo do gene GJB2, por sua vez, revelou a mutação 35delG em heterozigose na mãe do caso índice (I.2), na irmã mais velha ouvinte (II.4) e nos dois irmãos portadores de DA (II.6 e II.7). O pai (I.3), o caso índice (II.5) e a filha de sua meio-irmã (III.1) são homozigotos normais, portanto não apresentam a mutação 35delG; infelizmente, não pôde ser realizado o estudo molecular da meio-irmã (II.2) e do sobrinho (III.2). Os achados moleculares estão resumidos no heredograma da Figura 2.
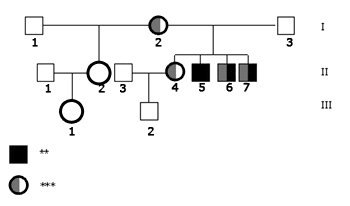
Figura 2. Heredograma da família mostrando os resultados da análise genética. - Legenda: ** Perda auditiva *** Heterozigoto 35delG N - Alelo Normal NR - Não Realizado.
Sabendo-se das restrições e problemas que a DA traz a seus portadores1 e da necessidade de seu diagnóstico o mais precoce possível, o que favorece a sua intervenção2,salienta-se aqui a importância de uma comunicação mais efetiva entre a fonoaudiologia e a genética.
O estudo audiológico completo, do núcleo familiar (pais e irmãos), complementado com o estudo da meio-irmã e dos sobrinhos, mostrou que os três irmãos tinham DA de características praticamente idênticas e que seus pais, irmã, meio-irmã e sobrinhos eram audiologicamente normais3-11.
O estudo mostra também que a DA encontrada no caso é de instalação pré-lingual e neurossensorial não-sindrômica12-14.
Isso permite que sejam propostos diversos mecanismos de herança15,16, sendo os mais prováveis o autossômico recessivo, no qual os pais normais não apresentam o fenótipo, são portadores heterozigotos e com recorrência do distúrbio na prole; poderia também ser um padrão de herança recessivo ligado ao cromossomo X, com mãe apresentando fenótipo normal, mas heterozigota portadora onde o distúrbio afetaria somente indivíduos do sexo masculino.
É possível, ainda, cogitar a respeito de herança autossômica dominante, com penetrância incompleta em um dos genitores ou decorrente de mutação em tecido germinativo de um deles15,16, e de herança materna, ou também chamada mitocondrial24, com manifestação clínica preferencial no sexo masculino, como ocorre, por exemplo, na neuropatia óptica de Leber.
O interessante da casuística ora descrita está relacionado às limitações do diagnóstico molecular nos indivíduos com deficiência auditiva neurossensorial não-sindrômica. Caso o estudo genético tivesse se limitado a um dos irmãos que se mostraram portadores da mutação 35delG (II.6 e II.7) e a seus pais, viriam à tona teorias que buscam explicar a presença dessa mutação em heterozigose em indivíduos com DA. De fato, uma das maiores dificuldades com relação ao aconselhamento genético de indivíduos portadores de mutações no gene da conexina 26, o que inclui a mutação 35delG, deve-se ao fato de que em aproximadamente 10 a 40% dos casos essas mutações são detectadas em apenas um dos alelos, e ainda assim resultando no fenótipo da perda auditiva. Em alguns casos essas mutações estão segregando com as deleções no gene GJB6, ou seja, o indivíduo é heterozigoto para mutações no gene GJB2 e no outro alelo apresenta uma das duas deleções envolvendo o gene GJB622,23. Na família em questão, entretanto, não foram encontradas as referidas deleções. Nos casos monoalélicos para mutações no gene GJB2 não é possível excluir a hipótese de interação de mutações na conexina 26 com mutações em outros genes relacionados à DA, podendo-se tratar de uma herança digênica. Outra hipótese para explicar tal situação seria a presença de genes modificadores, com efeito dominante negativo no alelo normal, o que nesse caso dependeria do background genético de cada indivíduo.
Se estamos ainda nos referindo a possibilidade de o indivíduo II.5 não ter se submetido ao diagnóstico molecular, qualquer geneticista trataria como a melhor possibilidade, uma situação onde não se explica a interação ocorrida no outro alelo e que nesse caso a mutação 35delG estaria envolvida no fenótipo de DA apresentado na família.
Desse modo, estariam excluídas as hipóteses de herança recessiva ligada ao X e assim, não sendo considerado, o risco de que a irmã (II.4) e a meia-irmã (II.2) viessem a gerar filhos do sexo masculino com DA, assim como o de herança mitocondrial, também desconsiderando o risco para a prole da irmã e da meio-irmã.
No entanto, o fato de o estudo também ter incluído todo o núcleo familiar permitiu que se verificasse que o fenótipo de DA segrega independentemente do genótipo 35delG. De fato, há dois indivíduos com a mutação e audiologicamente normais (I.2 e II.4) e, principalmente, um com DA, mas sem a mutação 35delG (II.5).
Desse modo, pode-se considerar o achado da mutação 35delG como casual, haja visto que essa mutação em heterozigose é muito comum na população normal, podendo chegar até a 4% em algumas populações17-21. Todas as hipóteses aventadas inicialmente em relação aos mecanismos de herança continuam válidas, impedindo o cálculo preciso do risco de recorrência nessa família e comprometendo, assim, o aconselhamento genético.
CONCLUSÃOEste trabalho evidencia a importância do diagnóstico interdisciplinar e a necessidade de crescente envolvimento da fonoaudiologia com a genética, aprimorando o diagnóstico etiológico e o aconselhamento genético nos casos de deficiência auditiva.
REFERÊNCIAS BIBLIOGRÁFICAS1. Godinho R, Keogh I, Eavey R. Perda Auditiva Genética. Rev. Bras. Otorrinolaringol. [online]. Jan./Fev. 2003, vol. 69, no. 1 [cited 06 Novembro 2005], p. 100-104. Available from World Wide Web: http://www.rborl.org.br/conteudo/acervo/print_acervo.asp?id=35. ISSN 1806-9312.
2. Azevedo MF. Avaliação audiológica no primeiro ano de vida. In: Lopes Filho, OC (org.). Tratado de Fonoaudiologia. São Paulo, Editora Roca Ltda.; 1997, cap. 11 p.239-63.
3. Russo ICP, Santos TMM. A Prática da Audiologia Clínica. São Paulo: Cortez editora; 2005.
4. Granato L, Pinto CF, Ribeiro MQ. Perda Auditiva de Origem Genética. In: Lopes Filho OC (org.). Tratado de Fonoaudiologia. São Paulo: Editora Roca; 1997, cap. 2 p. 25-53.
5. Pfeilsticker LN, Stole G, Sartorato EL, Delfino D, Maciel-Guerra AT. Genetic investigation of non-syndromic hereditary deafness. Rev Bras Otorrinolaringol 2004; 70: 182-6.
6. Petit C. Genes responsible for human hereditary deafness: symphony of a thousand. Nature Genet 1996:14:385-91.
7. Van Camp V, Willems PJ, Smith RJH. Non-syndromic hearing impairment: unparalleled heterogeneity. Am J Hum Genet 1997:60:758-64.
8. Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, DÁgruma L, et al. Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet 1998; 351: 394-8.
9. Antoniadi T, Rabionet R, Kroupis C,, Aperis GA, Economides J, Petmezakis J, et al. High prevalence in the Greek population of the 35delG mutation in the connexin 26 gene causing prelingual deafness. Clin Genet 1999; 55(5):381-2.
10. Green GE, Scott DA, McDonald JM, Woodworth GG, Sheffield VC, Smith RJ. Carrier rates in the midwestern United States for GJB2 mutations causing inherited deafness. JAMA. 1999 ; 281(23):2211-6.
11. Storm K, Willcox S Flothmann K, Van Camp G. Determination of the carrier frequency of the common GJB2 (connexin-26) 35delG mutation in the Belgian population using an easy and reliable screening method. Hum Mutat 1999; 14(3):263-6
12. Oliveira CA. Determinação da freqüência dos alelos 35delG no gene da conexina 26 em amostras da população brasileira. Campinas, 2005. [Tese de doutorado curso de Ciências Biomédicas, Faculdade de Ciências Médicas - UNICAMP].
13. Piatto VB, Bertollo EMG, Sartorato EL, Maniglia V. Prevalence of GJB2 mutations and the del(GJB6-D13S1830) mutation in Brazilian patients with deafness. Hearing Research. 2004; 196: 87-93
14. Del Castillo FJ, Rodriguez-Ballesteros M, Alvarez A, Hutchin T, Leonardi E, Oliveira CA, et al. A novel deletion involving the connexin-30 gene, Del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J Med Genet. 2005; 42(7):588-94.
15. Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu WQ, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non syndromic deafness. Nat Genet 1993; 4: 289-94.
16. Neto JFL, Pereira AC. O que há de Novo no Campo da Genética Molecular da Surdez: Descoberta de Genes para Surdez. Rev Bras Otorrinolaringol[online]. Mar/Abr 1999; (65)2 [cited 06 Outubro 2005], p. 106-113. Available from World Wide Web: http://www.rborl.org.br/conteudo/acervo/print_acervo.asp?id=1452. ISSN 1806-9312.
17. Sartorato EL. A genética da surdez. Pesquisa Fapesp, janeiro/fevereiro 2000, p.26-28.
18. Ramalho AS. As hemoglobinopatias hereditárias: Um problema de Saúde Pública no Brasil. Ribeirão Preto, Editora da Sociedade Brasileira de Genética; 1986. p. 119-28.
19. Faria I. Perda auditiva de origem genética: uma retrospectiva da literatura. São Paulo, 2001. [Monografia de Especialização em Audiologia clínica - Cediau].
20. Lloyd LL, Kaplan H. Audiometric interpretation: a manual of basic audiometry. Baltimore: University Park Press; 1978.
21. Carhart R. An improved method for classifying audiograms. Laryngoscope 1945;55:640.
22. Silman S, Silverman CA. Auditory Diagnosis - Principles and Applications. San Diego-London: Singular Publishing Group; 1997.
23. Lopes Filho OC, Carlos RC. Emissões Otoacústicas. In: Lopes Filho, OC (org.). Tratado de Fonoaudiologia. São Paulo: Editora Roca; 1997. cap.10 p. 221-37.
24. Manual do usuário - Ero Scan - Etymotic Research; 1999.
25. Manual do equipamento Biologic - Evoked Potential User Manual BIO -LOGIC; 1998.
26. Musiek FE, Lee WW. Potenciais auditivos de média e longa latência - In: Perspectivas Atuais em Avaliação Auditiva - Org. Musiek FE, Rintelmann WF. Barueri, São Paulo: Editora Manole; 2001. p.239-67.
27. McPherson DL. Late potentials of the auditory system (evoked potentials). San Diego: Singular Press; 1996.
1 Especialização em Audiologia. Instituto de Estudos Avançados da Audição, IEAA, Brasil. Fonoaudióloga.
2 Doutorado em Distúrbios da Comunicação Humana, Fonoaudiologia. Universidade Federal de São Paulo, UNIFESP, Brasil. Fonoaudióloga, Professora do Curso de especialização em Audiologia do Instituto de Estudos da Audição - IEAA.
3 Doutorado em Distúrbios da Comunicação Humana, Fonoaudiologia. Universidade Federal de São Paulo, UNIFESP, Brasil. Fonoaudióloga, Professora titular da PUC SP, Diretora do Instituto de Estudos Avançados da Audição - IEAA.
4 Doutorado em Genética e Biologia Molecular. Universidade Estadual de Campinas, UNICAMP, Brasil. Pesquisadora do Centro de Biologia Molecular e Engenharia Genética da UNICAMP, Livre Docente em Genética Médica pela UNICAMP.
5 Doutorado em Genética e Biologia Molecular. Universidade Estadual de Campinas, UNICAMP, Brasil. Médica Geneticista, Professora Titular do Departamento de Genética Médica da Faculdade de Ciências Médicas da UNICAMP.
6 Doutorado em Distúrbios da Comunicação Humana, Fonoaudiologia. Universidade Federal de São Paulo, UNIFESP, Brasil. Professor-Adjunto do Curso de Fonoaudiologia do Departamento de Fisioterapia, Fonoaudiologia e Terapia Ocupacional da Faculdade de Medicina da USP.
7 Bióloga. Bolsista de Capacitação Técnica FAPESP no Centro de Biologia Molecular e Engenharia Genética. IEAA - Instituto de Estudos Avançados da Audição
Endereço para correspondência: Flávia Maria Rodrigues Hoffmann - Rua Coronel Airton Gonçalves Froes 196 Jd. São Cristóvão Bragança Paulista SP 12906-040.
Tel. (0xx11) 4033-4066
Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) da RBORL em 17 de maio de 2007. cod.4530
Artigo aceito em 25 de fevereiro de 2008.