INTRODUÇÃOA Síndrome de Rendu-Osler-Weber ou Telangiectasia Hemorrágica Hereditária é uma rara displasia fibrovascular sistêmica, que tem como defeito básico uma alteração da lâmina elástica e camada muscular da parede dos vasos sangüíneos, o que os torna mais vulneráveis a traumatismos e rupturas espontâneas1,2.
A doença apresenta transmissão autossômica dominante, embora, em cerca de 20% dos casos, não exista histórico familiar. A sua incidência na população é de 1-2/100.000 e possui distribuição homogênea entre raça e sexo3.
O diagnóstico é feito seguindo os critérios de Curaçao: telangiectasias em face, mãos e cavidade oral; epistaxes recorrentes; malformações arteriovenosas com comprometimento visceral; histórico familiar. O diagnóstico é confirmado na presença de pelo menos 3 destas manifestações4.
As manifestações otorrinolaringológicas são as mais freqüentes, sendo a epistaxe recorrente a principal delas. Vasos sangüíneos de outras regiões também podem estar acometidos, principalmente pulmões, cérebro, pele e trato gastrointestinal1,4-6.
O tratamento é apenas paliativo, sem haver consenso a respeito da melhor opção terapêutica. O fundamental é promover o controle da doença pelo maior tempo possível. As opções variam desde tamponamentos anteriores e posteriores, cauterização química ou a laser das lesões até a tratamentos cirúrgicos, como a dermosseptoplastia e o fechamento da cavidade nasal pela técnica de Young5,6. Com o avanço dos procedimentos na área de Diagnóstico por imagem, é possível, hoje, realizar a embolização das artérias Maxilar, Etmoidal ou Esfenoidal, como também a ligadura das artérias Maxilar ou Palatina nos casos recorrentes, na tentativa de conter o sangramento e melhorar o quadro anêmico5.
Em virtude dessa falta de consenso a respeito da melhor terapêutica a ser empregada, todos os casos deveriam ser descritos para um maior conhecimento da doença. Também tem este trabalho o intuito de alertar os otorrinolaringologistas de que, frente a pacientes com epistaxes de repetição, deve-se fazer suspeita da Síndrome de Rendu-Osler-Weber e com o advento dos procedimentos endoscópicos, a realização de nasofibroscopia, que contribui de maneira importante no diagnóstico dessa síndrome.
Este estudo relata um caso de 1 paciente com a Síndrome de Rendu-Osler-Weber, atendido no ambulatório de Otorrinolaringologia da Faculdade de Medicina de Marília e revisa a literatura e tratamento disponíveis.
APRESENTAÇÃO DE CASO CLÍNICOC.L.S., de 53 anos procurou o Serviço de Otorrinolaringologia da Faculdade de Medicina de Marília referindo epistaxes de repetição em pequena intensidade há 15 anos, com intensificação dos quadros há três meses, chegando a apresentar sangramentos que persistiam por mais de uma hora três vezes ao dia, sempre precedidos de esforço físico ou espirros. Até então, não tinha tido necessidade de tamponamentos e/ou outros tratamentos.
Há cerca de 30 anos apresentava hemangiomas pelo corpo, principalmente em tórax, lábio inferior (Figura 1), língua, mucosa jugal, face, dedos e leito ungueal (Figura 2). Tinha como antecedente HAS, em uso de captopril 25mg 12/12h e propranolol 40mg 12/12h. Não fazia uso de medicação à base de salicilatos. O histórico familiar revelava pai com epistaxes de repetição, porém com quadros de intensidade mais leve e com lesões telangiectásicas em pele e mucosas e sem lesões viscerais e um neto com epistaxes leves aos 8 anos de idade.
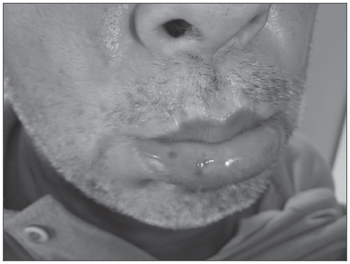
Figura 1. Telangiectasias em lábio inferior.
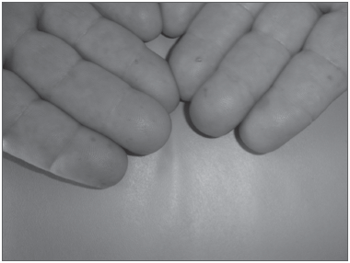
Figura 2. Telangiectasias em dedos de mãos.
Ao exame, foram evidenciadas lesões telangiectásicas disseminadas pelo tórax, unhas e dedos, lábio inferior, orofaringe, língua e face. À rinoscopia anterior, presença de pontos hemáticos em mucosa septal e de concha nasal média esquerda. Presença de crostas hemáticas em mucosa septal direita.
A radiografia de tórax e a ecografia abdominal e a tomografia computadorizada de crânio e de abdome não mostraram sinais de malformações arteriovenosas. Exames hematológicos mostraram Hb 8,3g/dl, Ht 26,6%.
Em um primeiro instante foi realizado tamponamento nasal, internação hospitalar e compensação hematológica do paciente com transfusão sangüínea. Após a retirada do tampão, com a recidiva das epistaxes, optou-se pela realização de cauterização das lesões nasais via endoscopia endonasal. Por fim, foi utilizada embolização maxilar direita com melhora da intensidade dos quadros de epistaxes, mas sem remissão completa. O paciente continua em acompanhamento ambulatorial neste serviço e foi encaminhado aos ambulatórios de Hematologia, Gastroenterologia e de Cirurgia torácica, obtendo alta dos últimos devido ausência de malformações arteriovenosas pulmonares e gastrintestinais. Ainda permanece em acompanhamento em ambulatório de hematologia para compensação de quadro de anemia e em uso de ácido aminocapróico como tratamento, desde então. O último exame hematológico mostrava Hb 12,4g/dL e Ht 37,4%.
REVISÃO BIBLIOGRÁFICA E DISCUSSÃO
HistóricoA primeira descrição da Doença foi feita por Henry Gawen Sutton, em 1864, que relatava um distúrbio que apresentava epistaxe, telangiectasias cutâneas e sangramentos internos7. Um ano após, Benjamin Guy Babington descreveu epistaxes de repetição em 5 gerações de uma família, sendo o pioneiro na observação do caráter hereditário da Doença4. Em 1876, John Wickham Legg também descreveu a doença, mas assim como Chiari em 1887 e Chaufffadin, em 1896, não foram capazes de diferenciar a Doença da Hemofilia2,4.
Somente em 1896, Louis Marie Rendu publicou a descrição de um homem com 52 anos com epistaxes recorrentes e com telangiectasias em pele de face e tronco, além de lesões em lábios e palato mole. Foi o primeiro a suspeitar de lesões nasais como causas de epistaxes. Notou, também, a presença de sangramentos nasais na mãe e em um irmão do paciente4.
Wiliam Osler descreveu em 1901 um relato de três casos onde descreveu a hereditariedade da doença, bem como foi o primeiro a relatar que vísceras poderiam ser afetadas4-6.
Em 1907, Frederick Parkes Weber fez uma descrição de uma série de casos, nos quais observou a presença de lesões em dedos, notadamente em sob as unhas. Em 1909, Hanes cunhou o termo Telangiectasia Hemorrágica Hereditária, porém a Doença é conhecida até hoje pelo epônimo de Síndrome de Rendu-Osler-Weber4.
Herança GenéticaÉ uma doença hereditária, com transmissão autossômica dominante, mas com cerca de 20% dos casos sem histórico familiar, podendo se tratar de mutações esporádicas7.
Ocorre em heterozigotos com penetrância incompleta em alguns casos. A forma homozigota é incompatível com a vida6. Muitos genes foram identificados e implicados na patogênese da doença, porém os mais importantes até o presente momento são o Endoglin (ENG) no cromossomo 9 e receptor da Activina A tipo II - Como I (ACVRL-1), sendo o primeiro associado a uma variante com manifestações pulmonares, conhecida por THH1 e o segundo com fenótipo mais suave e início tardio8. Os dois genes codificam uma glicoproteína de membrana que é expressa principalmente em células do tecido endotelial e constituem no receptor de superfície para o Fator de Crescimento ? (TGF-?) que mediará a remodelação vascular por meio de efeitos na produção de matriz extracelular8,9. Tanto o ENG, quanto o ACVRL - 1 e a função do TGF-? são essenciais à angiogênese8,9.
HistopatologiaOs achados histológicos, inicialmente descritos por Jahnke em 1970 como aumento de vasos submucosos em endotélio intacto, são dilatações das vênulas de capilares, pós-capilares e do tipo coletoras, com largas agregações alongadas de eritrócitos com canais de fibrina no tecido conectivo, células vermelhas espalhadas no interstício em volta dos vasos afetados, descontinuidade e degeneração endotelial2,5,10. O primeiro a descrever estas alterações foi Menefee et al., em 1975, após estudo com microscopia eletrônica10.
As células endoteliais descritas eram de três tipos: Normal - 90% delas; Degenerativas - 7%, com um denso citoplasma com menor quantidade de organelas. São menos aderentes às células adjacentes e desenvolvem fendas entre elas; Cubóides - 3% encontradas ao acaso10. A lâmina basal destas células mostrou-se intacta, por vezes reduplicada, mesmo onde as células estavam degeneradas ou havia fendas entre elas. Associada a reduplicação de lâmina basal, foi observado microfibrilas freqüentes em tecido conectivo, sem outras alterações evidentes2.
Braverman, em 1990, estudou o desenvolvimento das telangiectasias cutâneas. Em uma pele normal, as arteríolas estão conectadas às vênulas através de múltiplos capilares na região da derme papilar. Estes vasos vêm de outros vasos maiores da junção da derme com tecido adiposo. A ultra-estrutura normal da vênula pós-capilar inclui lúmen (L), células endoteliais e duas ou três camadas de células adventícias. Em um primeiro momento, as vênulas simples tornam-se dilatadas, mas ainda estão conectadas às arteríolas por meio de um ou mais capilares. Um infiltrado linfocítico perivascular está presente. Em um estágio mais avançado, as vênulas e seus ramos estão marcadamente dilatados, alongados e enrolados na derme. As arteríolas de conexão estão também dilatadas e esta dilatação faz com que os capilares desapareçam após algum tempo, propiciando malformações arteriovenosas (shunts e fístulas). O infiltrado linfocítico perivascular ainda está presente. As paredes dos vasos perdem suas fibras elásticas, enquanto que a camada endotelial, a membrana basal e a musculatura lisa permanecem intactas11.
DiagnósticoO diagnóstico clínico é feito com base nos Critérios de Curaçao estabelecidos pela divisão científica da Fundação Internacional de Telangiectasia Hemorrágica Hereditária. É baseado em:
1 - epistaxes - sangramentos nasais espontâneos e recorrentes;
2 - telangiectasias - múltiplas e em locais característicos (lábios, orofaringe, dedos e nariz)
3 - lesões viscerais - como telangiectasia gastrointestinal, com ou sem sangramentos, malformações arteriovenosas pulmonares, hepáticas, cerebrais e espinhais;
4 - história familiar - um parente de 1º grau com Telangiectasia Hemorrágica Hereditária4.
As múltiplas manifestações clínicas da Telangiectasia Hemorrágica Hereditária envolvem anormalidades vasculares nasais, cutâneas, pulmonares, cerebrais e de trato gastrointestinal5,6.
Manifestações Clínicas
NasaisEpistaxes causadas por sangramentos espontâneos de telangiectasias de mucosa nasal é a manifestação mais comum da Doença, mas não ocorre em todos os pacientes, cerca de 80% deles apresentam epistaxes recorrentes5. A severidade do quadro varia desde epistaxes tão severas que necessitam de múltiplas transfusões e suplementação oral de ferro a quadros tão brandos que a Doença nunca é suspeitada6. Os sangramentos iniciam-se por volta de 10 anos em alguns pacientes e até 21 anos em quase todos, porém tornam-se mais severos em décadas mais tarde em dois terços dos pacientes2.
CutâneasA lesão mais característica é a telangiectasia macular, com cerca de dois milímetros de diâmetro que ocorrem em face, lábios, nariz, língua, orelhas, mãos, tronco e pés2,5. Usualmente apresentam-se mais tardiamente que a epistaxe, sendo detectadas por volta da terceira década6. As lesões podem sangrar, porém o sangramento não é clinicamente importante6.
PulmonaresConsistem de malformações arteriovenosas através da comunicação direta da artéria e veia pulmonares por meio de um aneurisma de parede fina2. São múltiplos e aparecem em ambos os pulmões com predileção pelos lobos pulmonares inferiores. Estima-se que cerca de 60% das pessoas com malformações arteriovenosas pulmonares tenham Telangiectasia Hemorrágica Hereditária, mas que entre 15 e 33% das pessoas com a doença tenham malformações arteriovenosas pulmonares, embora esta incidência varie de acordo com o gene específico afetado9.
Produzem um shunt direita-esquerda6. Os sintomas iniciam-se por volta da terceira ou quarta década de vida e os pacientes podem, dependendo da importância do shunt, apresentar dispnéia profunda, fadiga, cianose ou policetemia6. Contudo, freqüentemente as manifestações iniciais são seqüelas de lesões neurológicas como acidente vascular cerebral isquêmico e abscesso cerebral.
Podem ser encontradas como um sopro extracardíaco holossistólico durante inspiração profunda ou por meio de exames de imagem6. A radiografia de tórax mostra uma massa típica com alargamento de artéria e veias, porém as lesões podem não ser encontradas pela sua localização póstero-inferior no pulmão2,5. Tomografia Computadorizada de tórax deve ser utilizada e angiografia pulmonar reservada para programação terapêutica de intervenção radiológica ou cirúrgica6.
CerebraisResultam de fístulas arteriovenosas pulmonares em cerca de 60% dos casos, malformações cerebrais em cerca de 28% e de medula espinhal em 8% e encefalopatia porto-sistêmica em 3%5,6.
Ocorrem entre 8 e 12% dos pacientes e os sintomas incluem: cefaléias, vertigens, síncopes, distúrbios visuais e auditivos, disartria, crises focais e generalizadas, obnubilação e paraparesias6.
Abscessos cerebrais, Acidentes Vasculares Cerebrais Isquêmicos, Encefalites Bacterianas ocorrem exclusivamente em pacientes com malformação arteriovenosa pulmonar e shunt direita-esquerda que facilitam a passagem de êmbolos sépticos para a circulação cerebral2.
Trato GastrointestinalOcorrem pequenos sangramentos recorrentes de trato gastrointestinal alto e baixo numa minoria substancial dos casos, em torno de 10%1. Ocorrem, predominantemente, na quinta ou sexta década de vida e são causados por telangiectasias mucosas similares as que ocorrem em mucosa oral e nasal1,4,5. Em metade dos pacientes a causa do sangramento pode estar localizada em estômago e duodeno5. Podem ocorrer telangiectasias maiores, angiodisplasia ou malformações arteriovenosas, menos comumente2. Há relatos de maior incidência de úlcera duodenal, porém sem correlações clínicas com a doença12.
O envolvimento hepático ocorre esporadicamente. Uma malformação arteriovenosa entre artéria e veia hepática pode produzir um shunt esquerda-direita, que pode levar a uma insuficiência cardíaca congestiva2,5. Um shunt entre as veias portal e hepática pode produzir encefalopatia hepática e sangramento gastrointestinal. Malformações entre artéria hepática e veia portal podem levar a hipertensão portal com varizes esofágicas. Podem ser suspeitadas quando há hepatomegalia ou um ruído sobre o fígado. Outros achados incluem elevação discreta de fosfatase alcalina e ?-GT, além de colestase. Ultra-sonografia e tomografia computadorizada podem ser utilizados para o diagnóstico6.
Outro tipo de envolvimento hepático é a cirrose da telangiectasia hemorrágica hereditária, caracterizada por vasos dilatados rodeados por uma quantidade de estroma variável e distribuído randomicamente pelo fígado. Entre estas estruturas, tecido hepático normal6.
TratamentoA terapia raramente é necessária para lesões cutâneas, mas quando as telangiectasias provocam distúrbios cosméticos ou sangram freqüentemente, podem ser utilizados a terapia a laser de ablação ou alguns agentes tópicos5.
Nas malformações pulmonares a terapia clássica é cirúrgica, com a desvantagem de ser um procedimento invasivo e de retirar a menor quantidade possível de tecido pulmonar são ao redor da malformação. Atualmente a terapêutica de escolha é a embolização dos vasos encontrados, embora faltem estudos mostrando o seguimento de pacientes a longo prazo9.
O tratamento de pequenos sangramentos gastrintestinais pode ser por meio de pequena dose combinada de estrogênio e progesterona, que segundo Carney13, reduziu a necessidade de transfusão mesmo após alguns meses depois de cessado o tratamento. Os resultados com laser via endoscópica e com coagulação bipolar são desanimadores, uma vez que as lesões, principalmente em intestino delgado, não são encontradas facilmente13. O tratamento com ácido aminocapróico tem sido sugerido, mas sua eficácia tem sido questionada13.
O tratamento das malformações hepáticas é conservador14. A experiência com ligação cirúrgica é limitada e os dados sobre a embolização, embora limitados, mostram uma mortalidade em cerca de 25% dos casos14.
Quanto às malformações em sistema nervoso, não há um consenso a respeito da melhor opção terapêutica entre tratamento conservador ou cirúrgico, ficando a decisão dependente dos riscos cirúrgicos e de sangramento letal, além da experiência do cirurgião e localização da lesão. Diferentes modalidades terapêuticas como a embolização e a radiocirurgia estereotática usando raios ? podem ser usados sozinhos ou em combinação6.
Por fim, este estudo enfatizará o controle dos quadros de epistaxe. Não há um tratamento definido como padrão1,15. As múltiplas modalidades variam desde tamponamentos anteriores e posteriores, cauterização elétrica e/ou química, ligadura dos vasos, dermosseptoplastia, terapia estrogênica, terapia hormonal e, mais recentemente, terapia com laser16. Porém nenhum tratamento obteve sucesso completo e por vezes são necessários mais de um tipo de tratamento para o controle do quadro de epistaxes1,2,16.
O tratamento tem por objetivo diminuir o número de episódios de sangramentos, bem como a sua intensidade16. Tenta reduzir a necessidade de transfusões sangüíneas e hospitalizações1,2. Objetiva, também, melhorar a qualidade de vida dos pacientes, bastante comprometida em virtude dos sangramentos de repetição principalmente1,2.
Em geral, ressalta-se que tratamentos com cauterização, pressão local ou tamponamentos são apenas paliativos, devendo ser usados em crises agudas1,2. Embolização arterial, terapia estrogênica ou com laser, procedimento de Young e cultura de lâminas epiteliais são utilizados como medidas terapêuticas definitivas em quadros leves, lembrando sempre que há grande recidiva de sangramentos e muitas vezes vários tratamentos são realizados16. Reservam-se a Dermosseptoplastia e os retalhos locais e microvasculares para os quadros mais graves16.
O tratamento hormonal é realizado com altas doses de estrógeno, o que induz a metaplasia da mucosa nasal para uma espessa camada de epitélio escamoso queratinizado que cobrem eventuais lesões nasais, protegendo-as assim de traumas locais2. Apesar de ter sua eficácia comprovada, a terapia hormonal não mais tem sido usada largamente devido aos potenciais efeitos colaterais, como náuseas, sensibilidade mamária, ganho de peso e doenças coronarianas6. Além disso, em pacientes masculinos pode haver ginecomastia, atrofia testicular e perda da libido1,2.
As cirurgias realizadas para o tratamento desta doença são a Dermosseptoplastia e o Fechamento das narinas pela técnica de Young16. A Dermosseptoplastia, ou cirurgia de Saunders, foi descrita em 1962 pelo mesmo e utilizava uma fina camada de pele para cobrir a área anterior do nariz. Podem ser retirados retalhos de pele de coxa ou braço, mucosa labial ou de membrana amniótica. A cirurgia consiste na retirada de toda mucosa afetada e a colocação do retalho sobre esta área, cobrindo septo nasal e cornetos inferiores bilateralmente17. É considerada o padrão ouro do tratamento1,2,6,13. O fechamento das narinas pela técnica de Young é realizado através de uma incisão na junção mucocutânea do vestíbulo nasal que são levantados retrogradamente. Apresenta sérios efeitos colaterais devido à respiração oral e é considerado um procedimento de última reservado para casos selecionados17.
A microembolização é realizada utilizando gelfoam ou substâncias esclerosantes a partir da artéria maxilar sob visualização de filmes angiográficos16. A braquiterapia e a injeção de cola de fibrina na região submucosa de septo nasal e de cornetos inferiores mostraram-se eficazes, porém com melhora temporária dos sintomas18. O tratamento com ácido aminocapróico é um tratamento pouco realizado, mas que possui melhora significante nos sintomas. Seus efeitos colaterais são náuseas, câimbras, diarréia, hipotensão, tonturas, rash cutâneo, miopatia, fadiga, rabdomiólise, disfunção renal e trombose16.
A terapia a laser é reservada para os quadros de sangramentos agudos na tentativa de contê-los. São descritos o laser de CO2, o Nd-YAG, o de argônio o KTP e o pulsed-dye laser, com comparação difícil entre eles devido a uma classificação não homogênea na literatura médica corrente mundial1,2,16.
COMENTÁRIOS FINAISA Telangiectasia Hemorrágica Hereditária é uma doença multissistêmica, que tem como primeira manifestação epistaxes de repetição. Desse modo, é de fundamental importância que o otorrinolaringologista esteja atualizado com relação à sua etiopatogenia e às opções terapêuticas, para que possa efetuar o diagnóstico correto, bem como prevenir complicações sistêmicas da doença.
Desse modo, esse trabalho teve o intuito de atualizar os conhecimentos sobre a moléstia, bem como relatar um caso clínico do ambulatório de Otorrinolaringologia da Faculdade de Medicina de Marília, uma vez que existem várias opções terapêuticas, mas ainda não há um tratamento inteiramente satisfatório, sendo de grande valia o registro de todos os casos encontrados a fim de comparar manifestações clínicas e condutas terapêuticas para que em um futuro próximo possamos obter uma padronização com resultados efetivos e uma melhora na qualidade de vida dos pacientes.
REFERÊNCIAS BIBLIOGRÁFICAS1. Rapoport PG, Uvo IP, Costa KS, Cecatto SB, Garcia RID. Síndrome de Rendu-Osler-Weber: tratamento clínico e cirúrgico. Rev Bras Otorrinolaringol 2003;694:577-80.
2. Maudonnet EN, Gomes CC, Sakano E. Telangiectasia Hemorrágica Hereditária (Doença de Rendu-Osler-Weber): um diagnóstico otorrinolaringológico. Rev Bras Otorrinolaringol 2000;662:172-80.
3. Pau H, Carney AS, Murty GE. Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): otorhinolaryngological manifestation. Clin Otolaryngol 2001;26:93-8.
4. Fuchizaki U, Miyamori H, Kitagawa S, Kaneko S, Kobayashi K. Hereditary Haemorrhagic Telangiectasia (Rendu-Osler-Weber Disease) Lancet 2003;362:1490-4.
5. Guttmacher AE, Marchuk DA, White RL. Hereditary Haemorragic Telangiectasia. New England J Med 1995;333: 918-24.
6. Haitjema T, Westermann CJJ, Overtoom TTC, Timmer R, Disch F, Mauser H, Lammers JWJ. Hereditary Hemorrhagic Telangiectasia (Osler-weber-Rendu Disease). Arch Intern Med 1996;56(8):714-9.
7. Carpes OLF, Moussalle MM, Ravanello R, Moraes VA, Swarowsky AM. Síndrome de Rendu-Osler-Weber: relato de caso e revisão bibliográfica. Rev Bras Otorrinolaringol 1999;654:354-6.
8. Dakeishi M, Shioya T, Wada Y, Shindo T, Otaka K, Manabe M, Nozaki J, Inoue S, Koizume A. Genetic epidemiolology oh Hereditary hemorragic telangiectasia in a local communityin the northern part of Japan. 2002;19(2):140-8.
9. Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, Simonneau G, Galie N, Loyd JE, Humbert M, NicholsWC, Morrel NW. Clinical and molecular genetic features of Pulmonary Hypertension in patients with Hereditary Hemorrhagic Telangiectasia. N Engl J Med 2001;345(5):325-34.
10. Menefee MG, Flessa HC, Gluck SPH. Hereditary Hemorragic Telangiectasia (Osler Weber Disease) - An electron microscopic study of the vascular lesions before and after therapy with hormones. Arch Otolaryngol 1975;101:246-51.
11. Braverman IM, Keh A, Jacobson BS. Ultrastructure and three-dimensional organization of the telangiectases of hereditary hemorrhagic telangiectasia. J Invest Dermatol 1990;95:422-7.
12. Perry WH. Clinical spectrum of Hereditary Hemorragic Telangiectasia (Osler Weber Disease). Am J Medicine 1987;82:899-97.
13. Carney PH, Murty GE. Hereditary haemorragic telangiectasia (Osler-WeberRendu syndrome): otorhinolaringological manifestations. Clin Otolaryngol 2001;26(2):93-8.
14. Garcia-Tsao G, Korzenik JR, Young L, Henderson KJ, Jain D, Byrd B, Pollack JS, White RI. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000;343(13):931-6.
15. Martins RHG, Nakajima V, Dias NH, Sousa JC. Epistaxe associada a Síndrome de Rendu-Osler-Weber (Telangiectasia Hemorrágica Hereditária). Arquivos de Otorrinolaringologia 1999;34.
16. Siegel MB, Keane WM, Atkins JP, Rosen MR. Control of epistaxis in patients with Herditary Hemorragic Telangiectasia. Otol Head Neck Surg 1991;105:675-9.
17. Lund VJ, Howard DJ. Closure of nasal cavities in the treatment of refractory Hereditary Haemorrhagic Telangiectasia. J Laryngol Otol 1997;111:30-3.
18. Turcato G, Pizzi GB, Polico R, Antonello M, Busetto M. Epistaxis in Rendu-Weber-Osler Disease. The role of Brachytherapy. Acta Otorhinolaryngol Ita 1996;16:513-6.
1 Médico Residente.
2 Doutor em Otorrinolaringologia, Chefe da Disciplina de Otorrinolaringologia da Faculdade de Medicina de Marília.
3 Mestre em Otorrinolaringologia, Docente da Disciplina de Otorrinolaringologia da Faculdade de Medicina de Marília.
4 Médica Otorrinolaringologista, Docente da Disciplina de Otorrinolaringologia da Faculdade de Medicina de Marília.
5 Médica Otorrinolaringologista, Docente Voluntária da Disciplina de Otorrinolaringologia da Faculdade de Medicina de Marília.
6 Médico Otorrinolaringologista, Ex-residente em Otorrinolaringologia da Faculdade de Medicina de Marília.
Disciplina de Otorrinolaringologia. Faculdade de Medicina de Marília.
Endereço para correspondência: Antônio José Cortez Juares - Rua Hidekichi Nomura 175 Fragata C Marília SP 17519-030.
Tel. (0xx14) 3402-1704 - Fax: (14) 3402-1655
Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) da RBORL em 3 de maio de 2005. Cod. 293.
Artigo aceito em 13 de setembro de 2005.
