INTRODUÇÃOA perda auditiva (PA) é a desordem sensorial mais comum1, sendo que a Perda Auditiva Sensorioneural (PASN) afeta aproximadamente 1 a 3 em cada 1000 recém-nascidos2. Estima-se que pelo menos 50% das perdas auditivas pré-linguais são causadas por alterações genéticas3, mas faltam dados epidemiológicos exatos sobre as perdas auditivas hereditárias pós-linguais1.
Os casos hereditários são divididos em sindrômicos e não-sindrômicos. Das perdas auditivas pré-linguais, 70% são não-sindrômicas e os 30% restantes são caracterizadas pela presença de outros sintomas e sinais (sindrômicas)1. Nas perdas auditivas não-sindrômicas, 80% são herdadas de modo autossômico recessivo (DFNB), 15 a 20% de modo autossômico dominante (DFNA) e menos de 2% ligado ao X (DFN) ou à herança mitocondrial2,3 (Figura 1).
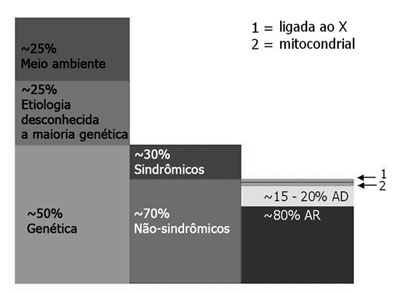
Figura 1. PASN pré-lingual: dados epidemiológicos em países desenvolvidos - A subdivisão da perda auditiva por causa (coluna 1), presença ou ausência de traços associados nos casos de etiologia genética (coluna 2) e o modo de herança no grupo não-sindrômico (coluna 3). Na coluna 3, a caixa 1 representa a perda auditiva ligada ao X (aproximadamente 1% das não-sindrômicas) e a caixa 2 a perda auditiva mitocondrial, que explica pelo menos 1%. AR, autossômica recessiva; AD, autossômica dominante. (Modificado de Schrijver, 2004)
A perda auditiva sindrômica caracteriza-se por manifestações adicionais, tais como: retinite pigmentosa (Síndrome de Usher), bócio eutiroideano e mal-formações da orelha interna (Síndrome de Pendred), anomalias renais (Síndrome de Alport) e presença de intervalo QT alongado (Síndrome de Jervell e Lange-Nielsen). Os sintomas e sinais associados são úteis quando podem ser observados, no entanto podem ser de aparecimento tardio ou não reconhecidos, tornando o diagnóstico incompleto4.
OBJETIVOPropor um roteiro para a investigação das PASN genéticas mais comuns, considerando os dados epidemiológicos, as informações e o desenvolvimento de novas tecnologias, as implicações clínicas e os aspectos bioéticos.
MATERIAL E MÉTODOSRealizada uma revisão criteriosa, utilizando os descritores: perda auditiva, sensorioneural, genética e diagnóstico, para compor um roteiro de investigação e de conduta.
REVISÃO DA LITERATURAA PASN genética pode seguir um padrão autossômico dominante, autossômico recessivo, ligado ao X ou de herança mitocondrial (Figuras 2 e 3). A base genética é altamente complexa. Mutações alélicas em alguns genes podem causar PA recessiva e dominante, mutações no mesmo gene podem causar PA sindrômica e não-sindrômica e, PA recessiva pode ser causada por uma combinação de duas mutações em diferentes genes do mesmo grupo funcional3,5.
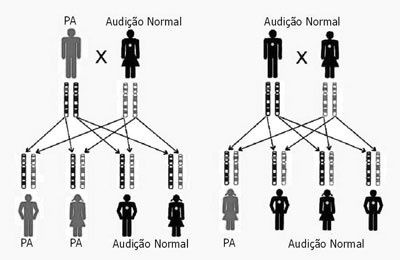
Figura 2. Modo de Herança - A primeira chave representa a herança de uma mutação Autossômica Dominante; uma faixa vermelha assinala uma mutação em um gene do pai. Na segunda chave está representada uma herança de uma mutação Autossômica Recessiva; a faixa vermelha representa uma mutação recessiva em um gene do pai e no mesmo gene da mãe; na forma dominante somente uma cópia é necessária para o indivíduo ser afetado e na forma recessiva as duas cópias de um mesmo gene precisam estar alteradas. (Modificado de REHM, 2003)
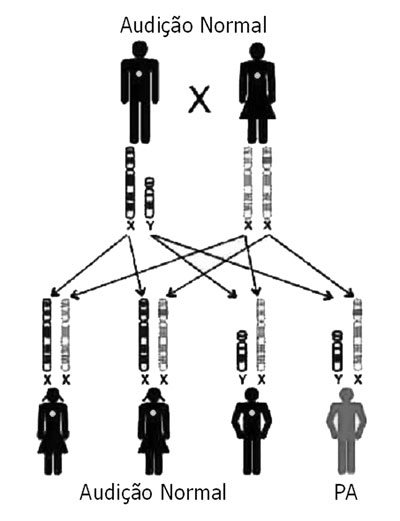
Figura 3. Modo de Herança - Esta chave representa uma mutação recessiva Ligada ao X; as filhas que herdam a cópia alterada da mãe não serão afetadas, pois receberam uma cópia normal do pai e os filhos têm 50% de chance de herdar o cromossomo X alterado da mãe. (Modificado de Rehm, 2003)
Um único gene, GJB2 (Gap Junction ?2), explica mais de 50% da perda auditiva recessiva. A Conexina 26 (Cx26), a proteína codificada pelo GJB2, pertence a uma família de proteínas de gap-junction responsável pelo transporte de íons, metabólitos e mensageiros secundários2. Estudos animais sugerem que Cx26 participa da reciclagem de íons potássio de volta para a endolinfa do ducto coclear, após a estimulação das células ciliadas sensoriais2. Esta perda auditiva não-sindrômica autossômica recessiva, no locus DFNB1, no cromossomo 13q11-12, caracteriza-se por ser congênita, tipicamente não-progressiva e de moderada a profunda3. O locus contém dois genes, GJB2 e GJB63,6,7.
-O POU3F4 codifica um fator de transcrição. A PA é ligada ao X, não-sindrômica, progressiva e profunda (DFN3) e pode apresentar um componente condutivo, pela fixação da platina. A TC pode ser útil, um alargamento do meato acústico interno ou uma dilatação entre o meato acústico interno e a orelha interna podem ser vistos. A pressão perilinfática está aumentada e a perilinfa da orelha interna pode jorrar durante a remoção cirúrgica do estribo3.
PAs SindrômicasSíndrome de Pendred: é responsável por 4 a 10% da perda auditiva hereditária pré-lingual no mundo3,4. Uma das formas mais comuns de PA sindrômica é uma desordem autossômica recessiva composta de PA e um defeito na organificação do hormônio tireoidiano, resultando em um bócio eutireoidiano. O bócio não está presente consistentemente e às vezes se manifesta só no adulto3. Alargamento do aqueduto vestibular foi encontrado em quase todos os pacientes e está associado a uma cóclea dismórfica, que apresenta 1,5 e não 2,5 voltas (Displasia de Mondini)3,4. A PA é caracteristicamente pré-lingual (não necessariamente congênita), sensorioneural ou raramente mista, de severa a profunda, freqüentemente estável, mas podendo ser flutuante e progressiva3,4. Mutações no gene SLC26A4, também conhecido como PDS, responde pela maioria, mas não por todos os casos de Síndrome de Pendred. Um teste laboratorial útil para diagnóstico é o teste da descarga do perclorato e podemos utilizar também a tomografia computadorizada38,9.
Síndrome de Usher: desordem autossômica recessiva, caracterizada por PA, perda progressiva da visão pela retinite pigmentar e em alguns casos anormalidades do equilíbrio. A síndrome é geneticamente heterogênea. Existem vários genes que podem causar a síndrome. Clinicamente está subdividida em: tipo 1, tipo 2, tipo 3 e atípica. A Síndrome de Usher tipo 1 caracteriza-se por PA congênita, severa a profunda, retinite pigmentar com início pré-puberdade e arreflexia vestibular. A PA no Usher tipo 1 e 2 é congênita, enquanto que a retinite pigmentar pode ser tardia e não notada até a adolescência4. A prevalência estimada é de 3 a 4,5 em 100.00010. O gene miosina VIIa é responsável pelo Usher tipo 1B; a miosina VIIa se expressa nas células ciliadas do Órgão de Corti e no vestíbulo; na retina a miosina VIIa está presente nas células epiteliais pigmentadas da retina10.
-Síndrome de Alport: decorre de alterações nas cadeias de colágeno tipo IV e os sintomas refletem o comprometimento da membrana basal de vários órgãos. A herança ligada ao X é predominante em 85% dos casos e a forma autossômica recessiva é responsável por 15% dos casos. Caracteriza-se por hematúria, evolui para falência renal e pode ser acompanhada por PASN e defeitos oculares. A incidência de Síndrome de Alport é descrita como 1 em 200 mil. A PA é um achado freqüente e um dos primeiros sintomas na síndrome, sendo um fator relevante para o prognóstico da evolução da doença renal. A PA é de intensidade variável, progressiva, bilateral e simétrica, acometendo as freqüências médias e altas. Na investigação das PASN nas crianças com hematúria, nos adolescentes e adultos masculinos em estágio final de falência renal e nos pacientes com história familiar de doença renal em irmãos ou parentes do lado materno, deve-se pensar em Síndrome de Alport. Devido à complexidade e ao alto custo, os métodos disponíveis para diagnóstico genético são restritos aos casos selecionados11-13.
Síndrome de Jervell e Lange-Nielsen: mutações no KCNQ1 OU KCNE1, herança autossômica recessiva, repolarização atrasada do canal potássio, com PASN congênita profunda, displasia cócleo-sacular (Scheibe), condução cardíaca anormal, intervalo QT prolongado e morte súbita. O problema cardíaco pode não ser diagnosticado, pois pode ser difícil detectá-lo1,4,14.
PAs MitocondriaisA PASN está presente em 42 a 70% dos indivíduos com desordens mitocondriais e pode ser não-sindrômica ou sindrômica. Mutações no DNA mitocondrial foram identificadas em aproximadamente 3% dos pacientes com PASN, sendo que as mutações são transmitidas exclusivamente através da mãe3. Entre os pacientes que recebem tratamento convencional com aminoglicosídeos (com níveis terapêuticos e por curto período), mais de 25% apresentam PASN e 50% destes são portadores da mutação 12S rRNA. A patogênese da PA genética mitocondrial baseia-se na alta necessidade de ATP nas células ciliadas da cóclea, sendo que a redução do ATP disponível, causada pela disfunção da fosforilação oxidativa mitocondrial devido às mutações, resulta em distúrbios do gradiente iônico na orelha interna. Mutações mitocondriais podem estar relacionadas com a PA progressiva da idade, a presbiacusia3.
A PASN mitocondrial pode ser sindrômica. Observada em: na Síndrome de Kearns-Sayre (oftalmoplegia progressiva e PA); na encefalopatia mitocondrial com acidose láctica e episódios de acidente vascular cerebral (MELAS) ou, na diabetes e PA de herança materna3.
Avaliação das Perdas Auditivas GenéticasTão logo se suspeite de PASN genética, deve ser feita uma história completa pré-natal, médica e familiar. O exame clínico e o genético são necessários para excluir características comuns à etiologia sindrômica ou infecciosa congênita. Um exame oftalmológico deve ser realizado, pois alterações oculares estão presentes em mais da metade das crianças com PA, de severa a profunda. Testes laboratoriais devem ser individualizados e dirigidos de acordo com a suspeita diagnóstica. TSH e teste da descarga do perclorato na suspeita de Síndrome de Pendred; testes de urina e função renal em crianças com possível Síndrome de Alport; um ECG para avaliar o intervalo QT nas Síndromes de Jervell e Lange-Nielsen. Estudos de imagem podem incluir TC de alta resolução para avaliar a presença de malformação de Mondini ou uma Ressonância Magnética para visualizar o nervo acústico, excluir aplasia ou destruição da orelha interna infecciosa, sendo especialmente importante antes de eventual cirurgia de implante coclear3. As audiometrias são componentes importantes do processo de avaliação15. Os testes genéticos têm implicações para todos os membros da família e a privacidade genética dos parentes do paciente deve ser considerada15. A história familiar pode estigmatizar membros da família, pode haver necessidade de se entrar em contato com familiares para a interpretação acurada de resultados, respeitando sempre o direito à privacidade e autonomia dos pacientes.
O espectro de testes moleculares disponíveis clinicamente ou em investigação está crescendo e pode ser obtido no website Genetests (http:// www.genetests.org/servlet/access)3,16-19.
DISCUSSÃOA história familiar detalhada é um dos mais importantes indícios da etiologia da PA, definindo o padrão de herança nesta família. É importante coletar detalhes sobre a saúde e a audição de irmãos, pais, avós e outros membros próximos da família. Investigar a ocorrência de consangüinidade e também a etnia da família. O exame físico criterioso do paciente pode identificar características de formas sindrômicas ou confirmar uma forma isolada (não-sindrômica).
Conforme comentado, os testes laboratoriais devem ser solicitados de acordo com a suspeita clínica. Os exames audiológicos são importantes para determinar o grau da perda auditiva, para acompanhar a PA nos casos evolutivos e também para orientar quanto à reabilitação dos pacientes. Deve-se comparar os resultados dos testes audiológicos do paciente com os exames de outros membros da família.
A avaliação do paciente com PA requer uma abordagem multidisciplinar e deve incluir aconselhamento e suporte aos pais, sendo que o aconselhamento genético visa otimizar a utilização dos recursos clínicos mais adequados.
A avaliação genética familiar é parte importante do processo diagnóstico e da solicitação dos testes genéticos específicos, em uma criança que apresenta alteração auditiva. Os pais devem receber informações sobre a causa e o comportamento da PA, implicações ou não de outros órgãos, a chance de recorrência em outros filhos e a possibilidade de afetar outros membros da família. Os testes genéticos são parte da avaliação, na tentativa de confirmar um diagnóstico específico. Deve-se informar sobre o diagnóstico e as condições que acompanham o quadro, o prognóstico, o modo de herança e as opções de tratamento. Se os testes genéticos resultarem em implicações importantes para a saúde dos parentes deve-se entrar em contato com eles. O aconselhamento genético pré e pós-teste é importante para que o paciente entenda as vantagens e as limitações de um teste genético particular, assim como as implicações dos resultados para ele e a família dele.
Os testes genéticos são parte da avaliação e devem contribuir para confirmar ou excluir um diagnóstico específico. A rápida introdução dos testes genéticos na prática clínica torna necessária a busca por informações sobre os riscos e os benefícios destes testes e também até que ponto a introdução do exame é útil e apropriada. A GJB2 (conexina 26) é a causa mais comum de PA não-sindrômica, portanto deve ser o primeiro passo na investigação da análise de mutação, após obtenção do consentimento esclarecido. Podem ser analisados o sangue periférico e células bucais obtidas com um swab. Os métodos empregados para identificar a mutação GJB2 são rápidos, relativamente econômicos, altamente sensíveis e específicos, mas limitados, pois o número de pontos de mutação investigado é baixo. Pode ser útil nos indivíduos com PA genética, de etiologia não estabelecida, sem outros traços aparentes e com estudos de imagem negativos (Figura 4).
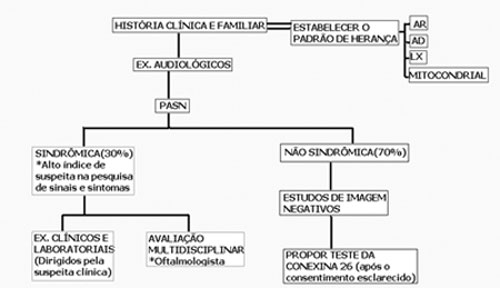
Figura 4. Screening da PASN genética - O otorrinolaringologista deve ter alto índice de suspeita para determinar as formas sindrômicas (evidências de síncope ou morte súbita em outros membros da família, nas Síndromes de Jervell e Lange-Nielsen; alargamento do aqueduto vestibular e teste da descarga do perclorato positivo na Síndrome de Pendred); os exames solicitados devem ser dirigidos pela suspeita clínica; mais de 50% das crianças com PASN de severa a profunda têm alterações oftalmológicas. Quando a PASN é isolada e os exames de imagem são negativos deve ser proposto o Teste da Conexina 26, respeitando a privacidade e a autonomia dos pacientes. AR: autossômica recessiva; AD: autossômica dominante; LX: ligada ao X; Mitocondrial: herança mitocondrial.
O diagnóstico da PASN, impulsionado pela genética e pelos biomarcadores, certamente crescerá nos próximos 10 anos; diagnóstico precoce, terapia dirigida e monitorização da doença substituirão o paradigma corrente de diagnóstico tardio e terapia11 (Figura 5).
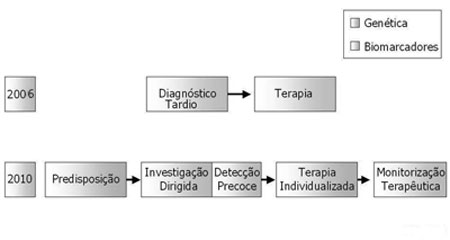
Figura 5. Diagnóstico da PASN - O diagnóstico da PASN será impulsionado pela genética e pelos biomarcadores. (Modificado de Bell, 2004)
-Os dados epidemiológicos estimam que pelo menos 50% das perdas auditivas pré-linguais são determinadas por alterações genéticas.
-As histórias clínica e familiar são extremamente importantes na elaboração do diagnóstico das PASN genéticas e contribuem para a determinação do padrão de herança.
-Através de um alto índice de suspeita, causas sindrômicas podem ser diagnosticadas ou excluídas, com uma cuidadosa avaliação e a base molecular da PA pode ser determinada mais seguramente do que antes.
-Os testes genéticos e a herança mitocondrial devem ser considerados em famílias com múltiplos indivíduos afetados, estando esta última afastada se houver nítida transmissão através de um homem.
-Nas PASN não-sindrômicas a análise de mutação GJB2 deve ser proposta.
REFERÊNCIAS BIBLIOGRÁFICAS1. Van Laer L, Cryns K, Smith RJH, Van Camp G. Nonsyndromic hearing loss. Ear Hear 2003;24:275-88.
2. Gürtler N, Kim Y, Mhatre A, Müller R, Probst R, Lalwani AK. GJB2 Mutations in the Swiss hearing impaired. Ear Hear 2003;24:440-7.
3. Schrijver I. Hereditary Non-syndromic sensorineural hearing loss. J Mol Diag 2004;6:275-84.
4. Friedman TB, Schultz JM, Ben-Yosef T, Pryor SP, Lagziel A, Fisher RA, et al. Recent advances in the understanding of syndromic forms of hearing loss. Ear Hear 2003;24:289-302.
5. Rehm HL. Genetics and the Genome Project. Ear Hear 2003;24:270-4.
6. Zelante L, Gasparini P, Estivill X, et al. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum Mol Genet 1997;6:1605-9.
7. Wilcox SA, Saunders K, Osborn AH, et al. High frequency hearing loss correlated with mutations in the GJB2 gene. Hum Genet 2000;106:399-405.
8. Coucke P, Van Camp G, Demirhan O, etal. the gene for Pendred syndrome is located between D7S501 and D7S692 in a 1.7-cM region on chromosome 7q. Genomics 1997;40:48-54.
9. Fugazzola L, Mannavola D, Cerutti N, et al. Molecular analysis of the Pendreds syndrome gene and magnetic resonance imaging studies of the inner ear are essential for the diagnosis of true Pendreds syndrome. J Clin Endocrinol Metab 2000;85:2469-75.
10. Martini A, Mazzoli M, Kimberling W. An introduction to the genetics of normal and defective hearing. Ann NY Acad Sci 1997;830:361-74.
11. Pirson Y. Making the diagnosis of Alport's syndrome. Kidney Int 1999;56:760-75.
12. Hudson BG, Tryggavason K, Sundaramoorthy M, Neilson EG. Mechanisms of the disease: Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Engl J Med 2003;348:2543-56.
13. Wester DC, Atkin CL, Gregory MC. Alport syndrome: clinical update. J Am Acad Audiol 1995;6:73-9.
14. Splawski I, Timothy KW, Vicent GM, et al. Molecular basis of the long QT syndrome associated with deafness. N Engl J Med 1997;336:1562-7.
15. Arnos KS. The implications of genetic testing for deafness. Ear Hear 2003;24:324-331.
16. Mafong DD, Shin Ej, Lalwani AK. Use of laboratory evaluation and radiologic imaging in the diagnostic evaluation of children with SNHL. Laryngoscope 2002;112:1-7.
17. Morzaria S, Westerberg BD, Kozak FK. Systematic review of the etiology of bilateral sensorineural hearing loss in children. Int J Pediatr Otorhinolaryngol 2004;68:1193-8.
18. Morzaria S, Westerberg BD, Kozak FK. Evidence-based algorithm for the evaluation of a child with bilateral sensorineural hearing loss. J Otolaryngol 2005;34:297-303.
19. Van Camp G, Smith RJH. Hereditary hearing loss homepage. Disponível na internet http://webhost.ua.ac.be//hhh Acessado em 16 junho de 2006.
20. Bell J. Predicting disease using genomics. Nature 2004;429:453-6.
1 Doutoranda em Medicina - Concentração em ORL - FCMSC -SP; Mestre em Medicina -Concentração em ORL - FCMSC - SP, Preceptora da Clínica de ORL - HSPM - SP.
2 Doutor em Medicina - Área de Concentração em Otorrinolaringologia, pela UNIFESP - EPM, Médico Chefe de Clínica Adjunto da Santa Casa de Misericórdia de São Paulo.
Faculdade de Ciências Médicas da Santa Casa de São Paulo.
Endereço para correspondência: Dra. Fatima Regina Abreu Alves - Av. Moema 801 São Paulo SP 04077-023.
Tel.(0xx11) 5052-4344 / 5052-0093.
Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) da RBORL em 19 de novembro de 2005. cod. 1589.
Artigo aceito em 19 de junho de 2006.
