INTRODUÇÃOA anemia de Fanconi (McKusick 227650), também conhecida como pancitopenia hereditária, é uma síndrome caracterizada por distúrbios hematológicos associados a malformações congênitas, de herança autossômica recessiva, com expressão clínica variável. Entre as manifestações clínicas mais características incluem-se o atraso do crescimento, polegares hipoplásicos (ou ausentes) e rádio curto (ou ausente). Ao lado de outras afecções hereditárias como a síndrome de Bloom e a ataxia-telangiectasia, a anemia de Fanconi faz parte do grupo de síndromes marcadas pela instabilidade genômica, o que predispõe os pacientes a diferentes tipos de câncer. A instabilidade genômica manifesta-se, no estudo citogenético, como quebras e rearranjos em diferentes cromossomos.
O tratamento da anemia de Fanconi baseia-se em medidas de suporte, no uso de medicamentos (andrógenos, corticosteróides, fatores de estimulação de colônias) e, em casos selecionados, no transplante alogênico de medula óssea. A maior parte dos pacientes morre em conseqüência de complicações hematológicas ou sépticas relacionadas à pancitopenia. Por outro lado, aqueles que desenvolvem neoplasias malignas, além de não serem bons candidatos ao tratamento cirúrgico, são mais propensos aos efeitos adversos da radioterapia e da quimioterapia, de modo a dificultar ainda mais o planejamento terapêutico.
A seguir, faz-se uma revisão sumária da literatura e relata-se um caso de carcinoma de células escamosas da hipofaringe numa jovem com anemia de Fanconi.
REVISÃO DA LITERATURAInicialmente descrita em 1927, a anemia de Fanconi é um raro distúrbio autossômico recessivo, genotípica e fenotipicamente heterogêneo, caracterizado por anemia aplásica constitucional, malformações congênitas, instabilidade cromossômica e predisposição ao desenvolvimento de neoplasias.1,2 As manifestações clínicas são bastante variadas e incluem: retardo no crescimento e na maturação sexual, baixa estatura, anormalidades esqueléticas (rádio e polegar), aplasia de medula óssea, manchas café-com-leite e malformações cardíacas e renais. O diagnóstico é feito por meio de estudo citogenético, que demonstra uma maior tendência à ruptura cromossômica por agentes alquilantes como a mitomicina C e o diepoxibutano.
A base genética da anemia de Fanconi é tão complexa quanto a grande variação na apresentação clínica. Pelo menos oito subtipos (A, B, C, D1, D2, E, F e G), cada qual com um defeito genético distinto, já foram identificados. A patogênese está relacionada a uma falha nos mecanismos de reparo do DNA, com o conseqüente acúmulo de uma grande quantidade de mutações. Estudos recentes sugerem que os genes da anemia de Fanconi, assim como os genes BRCA1 e BRCA2, atuam numa via final comum, que se relaciona com o reparo do DNA.3
Em virtude da incapacidade de manter a integridade genômica, observa-se um alto grau de instabilidade cromossômica. Os pacientes possuem um risco aumentado de desenvolver neoplasias, que se acentua com o aumento da expectativa de vida.4 O tipo mais prevalente é a leucemia (principalmente mielóide aguda), seguindo-se, em ordem decrescente, os carcinomas de células escamosas (trato aerodigestivo alto, ânus, genitália e pele) e as neoplasias hepáticas, cerebrais e renais.5 As neoplasias manifestam-se em geral por volta dos 16 anos, não sendo incomum, com o avançar da idade, o aparecimento de outros tumores na mesma pessoa.6 Ademais, relatam-se casos em que o diagnóstico do câncer precedeu o diagnóstico da síndrome.
O tratamento dos tumores sólidos associados à anemia de Fanconi deve ser preferencialmente cirúrgico, mesmo com um risco operatório aumentado em decorrência da aplasia de medula óssea.7 Os carcinomas epidermóides de cabeça e pescoço costumam ser biologicamente mais agressivos, de modo a exigir uma terapia adjuvante. No entanto, por causa dos defeitos nos mecanismos de reparo do DNA, os pacientes são muito suscetíveis aos efeitos colaterais da radioterapia e quimioterapia, em especial a mielossupressão. Mesmo assim essas modalidades terapêuticas devem ser consideradas quando a intervenção cirúrgica for contra-indicada. Nesses casos, recomenda-se o uso de doses menores que as habituais, regimes individualizados (que não incluam agentes alquilantes ou cisplatina) e o uso de agentes radioprotetores como a amifostina.8-10
RELATO DO CASOTrata-se de uma mulher de 24 anos, de cor parda, solteira, estudante, que, após receber, aos 12 anos, o diagnóstico de anemia de Fanconi, passou a fazer uso regular de medroxiprogesterona e oximetolona. Havia mais dois casos diagnosticados na família: uma irmã de 21 anos e um irmão que faleceu aos 15 anos em conseqüência de leucemia. Havia, também, história de consangüinidade entre os pais.
A paciente, que não era etilista nem tabagista, foi internada em um hospital universitário por causa de rouquidão, disfagia, odinofagia e perda ponderal, de caráter progressivo, de seis meses de evolução. O exame clínico inicial evidenciou hipodesenvolvimento ponderal (peso= 28,7 kg) e estatural (altura= 1,32 m), manchas café-com-leite no tronco e desnutrição. O hemograma demonstrou pancitopenia acentuada: hemoglobina 5,9 g/dl, leucócitos 800/mm3 e plaquetas 29.100/mm3. A videolaringoscopia revelou lesão vegetante e ulcerada localizada, em sua maior extensão, no seio piriforme esquerdo, com invasão do esôfago proximal (Figura 1). Uma tomografia computadorizada da região cervical mostrou, na região da hipofaringe, massa com densidade de partes moles acometendo o seio piriforme esquerdo e estendendo-se desde as pregas ariepiglóticas até o nível das cartilagens aritenóides, com provável espessamento parietal concêntrico, segmentar, do esôfago cervical. Não se observaram linfonodomegalias (Figura 2). A biópsia da lesão revelou carcinoma de células escamosas, invasor e ulcerado, moderadamente diferenciado.
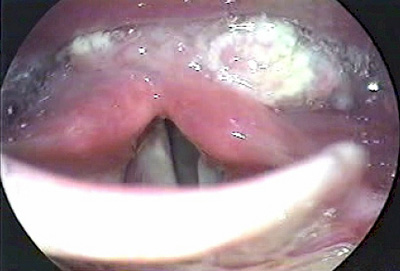
Figura 1. Aspecto do tumor à videolaringoscopia: lesão vegetante acometendo, em sua maior extensão, o seio piriforme esquerdo.
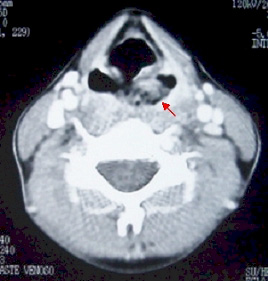
Figura 2. Tomografia computadorizada da região cervical, corte axial: massa na região do seio piriforme esquerdo.
Durante a internação, a paciente apresentou vários episódios de febre na vigência de neutropenia, o que motivou a prescrição de diversos antimicrobianos em associação (ceftazidima, amicacina, imipenem, vancomicina, anfotericina B e fluconazol).
A possibilidade de tratamento cirúrgico (faringoesofagectomia total com interposição de tubo gástrico retroesternal, esvaziamento cervical bilateral, tireoidectomia parcial e radioterapia pós-operatória) foi descartada em virtude das precárias condições clínicas e de recusa da paciente. A quimioterapia foi contra-indicada pela Clínica Oncológica, que recomendou radioterapia paliativa.
Coincidindo com a radioterapia houve recrudescimento da disfagia e da odinofagia, bem como reaparecimento da febre, o que motivou a sua interrupção (dose total de radiação recebida: 19,8 Gy). A paciente desenvolveu processo pneumônico em vigência de grave neutropenia (leucócitos 100/mm3), vindo a falecer apesar do tratamento antimicrobiano instituído.
DISCUSSÃOEm expressiva porcentagem dos casos, os carcinomas de células escamosas da hipofaringe estão intimamente relacionados ao uso do tabaco e do álcool, acometendo em geral pessoas acima dos 60 anos, com predomínio do sexo masculino.11 No caso ora relatado - uma jovem de 24 anos, sem história de etilismo ou tabagismo -, somente a concomitância de um outro fator predisponente como a anemia de Fanconi poderia explicar o surgimento desse tipo de neoplasia. A propósito, num estudo em que se avaliaram 754 pacientes acometidos pela síndrome, observou-se, em relação à população geral, um aumento de aproximadamente 500 vezes da incidência de tumores da cabeça e do pescoço, com nítido predomínio do sexo feminino (2:1) e uma idade média, quando do diagnóstico, de apenas 31 anos.12
COMENTÁRIOS FINAISTendo em vista a grande variação da apresentação clínica, o pequeno número de casos registrados na literatura e a elevada morbidade das diferentes formas de tratamento, reveste-se de grande dificuldade a abordagem das neoplasias de cabeça e pescoço associadas à anemia de Fanconi. Por conseguinte, todo esforço deve ser feito no sentido de sua detecção precoce, por meio de exame médico periódico que deve incluir, obrigatoriamente, uma cuidadosa avaliação otorrinolaringológica.
REFERÊNCIAS BIBLIOGRÁFICAS 1. Fanconi G. Familiäre infantile perniziösaartige Anämie (perniziöses Blutbild und Konstitution). Jahrb Kinderheilkund 1927; 117:257-80.
2. Alter BP, Young NS. The bone marrow failure syndromes. In: Nathan DG, Orkin SH, eds. Hematology of infancy and childhood. Philadelphia, PA: Saunders; 1998:237-335.
3. Venkitaraman AR. A growing network of cancer-susceptibility genes. N Engl J Med. 2003; 348(19):1917-9.
4. Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood 2003;101:822-6.
5. Kutler DI, Singh B, Satagopan J, Batish SD, Berwick M, Giampietro PF, et al. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood 2003;101:1249-56.
6. Alter BP. Cancer in Fanconi anemia, 1927-2001. Cancer 2003;97:425-40.
7. Doerr TD, Shibuya TY, Marks SC. Squamous cell carcinoma of the supraglottic larynx in a patient with Fanconis anemia. Otolaryngol Head Neck Surg 1998;118(4):523-5.
8. Alter BP. Radiosensitivity in Fanconis anemia patients. Radiother Oncol 2002;62(3):345-7.
9. Bremer M, Schindler D, Gross M, Dork T, Morlot S, Karstens JH. Fanconis anemia and clinical radiosensitivity report on two adult patients with locally advanced solid tumors treated by radiotherapy. Strahlenther Onkol 2003;179(11):748-53.
10. Momm F, Bechtold C, Rudat V, Strnad V, Tsekos A, Fischer K et al. Alteration of radiation-induced hematotoxicity by amifostine. Int J Radiat Oncol Biol Phys 2001;51:947-51.
11. Vieira MBM. Tumores da hipofaringe. In: Campos CAH, Costa HOO, eds. Tratado de Otorrinolaringologia. Vol. 4: Cabeça e pescoço - Laringologia e voz. São Paulo: Roca; 2002. p.78-88.
12. Kutler DI, Auerbach AD, Satagopan J, Giampietro PF, Batish SD, Huvos AG, et al. High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Arch Otolaryngol Head Neck Surg 2003;129:106-12.
1 Médico Residente de Clínica Médica do Hospital das Clínicas da Universidade Federal de Minas Gerais - UFMG.
2 Médico Residente de Otorrinolaringologia do Hospital das Clínicas da Universidade Federal de Minas Gerais - UFMG.
3 Professor Adjunto do Departamento de Clínica Médica da Faculdade de Medicina da Universidade Federal de Minas Gerais - UFMG.
4 Professor Adjunto do Departamento de Otorrinolaringologia da Faculdade de Medicina da Universidade Federal de Minas Gerais - UFMG.
5 Professora Adjunta do Departamento de Pediatria da Faculdade de Medicina da Universidade Federal de Minas Gerais - UFMG / Doutora em Genética.
Endereço para correspondência: Rua Montes Claros 1124/102 - Anchieta Belo Horizonte MG 30310-370.
Tel. (0xx31) 3225-7733 /99686001 - E-mail: henriquelinshorta@yahoo.com.br
Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) da RBORL em 11 de junho de 2006. cod. 2109.
Artigo aceito em 2 de agostos de 2006.
