INTRODUÇÃOEm países desenvolvidos, estima-se que 60% dos casos de surdez pré-lingual têm bases genéticas, os outros 40% restantes estão entre as mais diversas etiologias (Marazita et al., 1993, Mustafa et al., 2001). Além disso, 1/1000 crianças torna-se surda após a aquisição da linguagem, ou seja, no período pós-lingual, antes de alcançar a idade adulta entre 30-50 anos, 0,3% da população manifesta perda auditiva acima de 65 decibéis (dB) e 2,3% da população manifesta entre 60 e 70 anos de idade. A prevalência continua a aumentar e alcança índices de 50% em octogenários (Paparella et al., 1989; Morton et al., 1991).
No Brasil, os fatores ambientais (67%) estão entre as principais etiologias da perda auditiva, com predomínio dos casos de rubéola congênita e anóxia neonatal (33,5%), seguindo-se aqueles de etiologia indefinida (18,5%) e os de herança autossômica recessiva (15,5%). Consideram-se neonatos com alto risco para surdez aqueles que têm entre 2-5% de chance de desenvolver perda auditiva se sofrerem um ou mais desses fatores seguintes: asfixia ou anóxia, com apgar abaixo de 7,1; meningite bacteriana, principalmente a causada por Haemophillus influenza; infecções congênitas pré-natais (sífilis toxoplasmose, rubéola, citomegalovírus, herpes); malformações de cabeça e pescoço; bilirrubina elevada (níveis que necessitem transfusão); história familial de perda auditiva; permanência em UTI neonatal por um período maior que 48 horas; peso de nascimento menor que um quilo e meio e administração de drogas ototóxicas à gestante ou no período neonatal (Simões et al., 1992).
Casos de surdez tardios, ou seja, pós-linguais, podem ocorrer também com o contato com drogas ototóxicas, história familial de perda auditiva, e em alguns casos pode ser uma perda auditiva súbita, de origem indefinida, sendo considerada idiopática.
Apesar da terapia gênica ainda estar muito distante em sua aplicação na prática terapêutica, o implante coclear é um recurso viável e acessível para indivíduos com perda auditiva bilateral severa e/ou profunda, que não se beneficiam do aparelho de amplificação sonora individual (AASI).
Nos casos onde a perda auditiva ocorreu antes da aquisição da linguagem (pré-lingual) um dos fatores mais importantes é a idade. Em crianças muito jovens os resultados são melhores do que em crianças maiores. Geralmente a causa desta perda auditiva tão precoce é congênita, de origem genética, sem lesão física do aparelho auditivo, o que favorece o implante coclear.
A mutação 35delG no gene da conexina 26 não é rara; pelo contrário, sua presença em heterozigose pode ser encontrada em até 3% dos indivíduos em algumas populações. Na Itália, porém, ela está em torno de 1:32, em Portugal é de cerca de 1:40, e na Espanha 1:45. Se forem agrupadas essas três populações européias, das quais descende boa parte da população brasileira, verifica-se que a freqüência média de heterozigotos para a mutação 35delG é de 1:42; considerando uma união aleatória de heterozigotos e a chance de 25% de descendentes afetados, temos que nessas regiões 1 a cada 5.069 crianças nasceriam surdas por homozigose da mutação 35deIG (Gasparini et al., 2000).
Os recentes progressos obtidos na biologia molecular, com a descoberta de diferentes genes envolvidos na perda auditiva, vêm possibilitando a identificação da etiologia da surdez. A alta prevalência de mutações no gene GJB2 e a sua facilidade de estudo possibilitam o diagnóstico de muitos pacientes e têm sugerido que esses indivíduos seriam candidatos potenciais ao implante coclear.
A mutação mais freqüente neste gene é a chamada 35delG, a maior envolvida nos casos de surdez com etiologia genética. Essa mutação é a perda de uma base guanina da seqüência de DNA deste gene na posição 35. A pesquisa da mutação 35delG na etiologia da surdez é muito importante, pois 2 a 4% dos indivíduos são portadores desta mutação, ou seja, são heterozigotos. A mutação 35delG corresponde de 75 a 80% das mutações possíveis de serem encontradas neste gene (Cohn, et al., 1999a; Cohn et al., 1999b).
É necessário que o indivíduo herde dois alelos mutados, sendo um do pai e outro da mãe, para que se expresse a surdez. Sendo assim, há a impossibilidade de a conexina 26 ser codificada pelo gene GJB2 alterado. Quando o paciente apresentar a mutação 35delG em heterozigose, significa que há mutação em apenas um dos alelos, sendo possível que o outro alelo codifique a proteína. Isso implica em um menor numero de conexina 26 codificada.
A proteína conexina 26 é indispensável ao funcionamento normal do ouvido interno, sendo as alterações no gene responsável pela sua codificação a principal causa de surdez pré-lingual não-sindrômica hereditária, o que é identificado pela pesquisa da mutação 35delG através do "teste de surdez genética".
Alguns estudos concluíram que o implante coclear em pacientes com perda auditiva relacionada ao gene GJB2 obtém igual ou melhor discriminação de fala comparada a crianças surdas pré-linguais, com surdez de etiologia não-esclarecida ou mesmo com surdez congênita, como por exemplo, por citomegalovírus (Sinnathuray et al., 2004; Green et al., 2002). Isso porque, quando o dano ocorre no gene, a expressão disso não interfere na colocação do implante, ou seja, a cóclea não está comprometida como no caso de surdez de causa traumática ou infecciosa como a meningite ou ototoxicidade, que lesa a estrutura física da cóclea, impedindo muitas vezes que um número maior de eletrodos seja introduzido, pela cóclea estar calcificada, por exemplo. Os danos genéticos são com relação à estrutura dos conexons, ocorrendo uma dificuldade de comunicação intercelular, o que não compromete a estrutura física da cóclea, favorecendo um implante com sucesso.
A viabilidade e os benefícios de rastreamento de mutações no gene da conexina 26 estão se refletindo na saúde pública. O uso de testes moleculares, em conjunto com os audiológicos, ajudará na detecção precoce da surdez, o que é de extrema importância no manejo desses pacientes, em particular nos casos de surdez progressiva, pois a estimulação da linguagem em seu período crítico faz com que as crianças aprendam a se comunicar antes que a surdez se torne mais grave. Além disso, é possível hoje, inclusive, o diagnóstico preditivo naqueles indivíduos afetados por mutações no gene da conexina 26, ainda sem manifestação da surdez. As conseqüências dessa predição, no âmbito social e familial, são enormes, seja em relação à prevenção da surdez, seja no auxílio e redução de custos destinados à educação especial desses indivíduos, seu tratamento médico e decisão profissional (Sobe et al., 2000; Sartorato et al., 2000).
O objetivo do presente trabalho foi averiguar a incidência da mutação 35delG em crianças candidatas e submetidas ao implante coclear que tiveram a surdez diagnosticada como supostamente idiopática.
MATERIAL E MÉTODOEsse estudo foi realizado no Ambulatório de Otorrinolaringologia do Hospital das Clínicas, UNICAMP-SP, setor de Implante Coclear e no Laboratório Genética Humana - CBMEG, UNICAMP-SP.
Foram avaliadas 32 crianças candidatas e usuárias de implante coclear, apresentando perda auditiva neurossensorial severa a profunda bilateral de origem supostamente idiopática. O principal critério de inclusão destas crianças no setor de Implante Coclear é o não-benefício do aparelho de amplificação sonora individual (AASI). Esses indivíduos foram submetidos a uma avaliação multiprofissional, entre eles otorrinolaringologistas, fonoaudiólogas, psicólogas, assistente social, enfermeiros e geneticistas.
O rastreamento da mutação 35delG foi feito a partir da extração de DNA do sangue periférico coletado em tubo Vacutainer de tampa roxa. Para a detecção da mutação 35delG foi utilizada a técnica de PCR alelo-específico (AS-PCR), usando primers e condições da reação em cadeia da polimerase previamente descritas, modificação patenteada pelo CBMEG - UNICAMP. (Oliveira et al., 2002). Este método discrimina facilmente o alelo normal do mutante e através de duas reações é possível distinguir homozigotos normais, homozigotos 35delG e heterozigotos portadores da mutação 35delG.
Os resultados da análise molecular foram correlacionados com a etiologia da perda auditiva apresentada pelos pacientes analisados.
RESULTADOSNesse estudo, 32 crianças com surdez idiopática foram submetidas ao rastreamento molecular da mutação 35delG no gene GJB2. A mutação foi encontrada em homozigose em 4 do total de crianças avaliadas (12% dos casos), sendo esta a causa da perda auditiva (gráfico 1). Em 6 crianças, a mutação foi encontrada em heterozigose (19% dos casos), não sendo possível diagnosticar a surdez desses pacientes. A mutação 35delG em heterozigose não diagnostica a causa da surdez, apenas comprova que o paciente é portador dessa mutação, e como apresentado na literatura, os pacientes heterozigotos são, via de regra, ouvintes. No gráfico abaixo podemos observar os resultados obtidos no estudo genético.
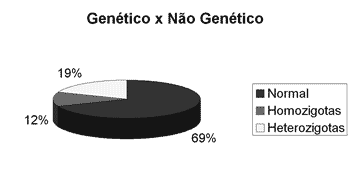
Gráfico 1.
Com o avanço das pesquisas nessa área, ficou evidente a importância dos estudos de mutações no gene GJB2, devido à facilidade de detecção de mutações na conexina 26, em particular a mutação 35delG, que é considerada a mutação mais freqüente em qualquer gene já estudado em caucasóides. Este é o primeiro gene indicado para análise molecular em famílias que apresentam perda auditiva sensorineural (Sobe et al., 2000).
Em estudo prévio realizado em uma amostra da população brasileira, mutações no gene GJB2 foram encontradas em 22% das famílias com surdez sensorineural não-sindrômica, indicando mais uma vez que a análise molecular desse gene em pacientes com perda auditiva não-associada a quadros sindrômicos deve ser o primeiro passo na determinação das causas de perda auditiva em nosso país (Oliveira et al., 2001). Isto é particularmente verdadeiro para os casos familiais, entre os quais a freqüência de encontro de mutações nesse gene foi de 50%, mas também para os casos esporádicos, entre os quais a freqüência foi de pouco mais de 11% (aproximadamente 1:9) (Oliveira et al., 2002). No Brasil, foi determinada a prevalência de 0,97% de portadores da mutação 35delG, aproximadamente 1:103 heterozigotos, em um rastreamento realizado em 620 neonatos, na região de Campinas-SP (Oliveira et al., 2004).
Nossos resultados são concordantes com estudos realizados e descritos na literatura em várias populações. A relativa distribuição da mutação 35delG para a perda auditiva não-sindrômica nessas populações variou de 0% (Omã, Coréia e Japão) a 70% (Itália, Espanha e Grécia), demonstrando a heterogeneidade genética existente entre os diversos paises, apesar de alguns desses estudos terem sido baseados em pequeno número de pacientes, além de critérios de investigação da perda e os métodos de rastreamento da mutação terem sido diferentes entre os mesmos (Liu et al., 2002).
CONCLUSÃONo presente estudo, os dados obtidos confirmaram a alta prevalência da mutação 35delG no gene GJB2 em casos de perda auditiva neurossensorial não-sindrômica bilateral profunda. E foi possível, também, diagnosticar como genética a causa da surdez em uma parcela significativa de crianças.
Os resultados reforçam a importância do estudo molecular em pacientes com surdez de origem supostamente idiopática, uma vez que esse exame possibilita esclarecer a etiologia da perda auditiva.
REFERÊNCIAS BIBLIOGRÁFICAS 1. Cohn ES, Kelley PM, Fowler TW et al. Clinical studies of families with hearing loss attributable to mutations in the connexin gene (GJB2/DFNB1). Pedriatric 1999a;103(3):546-50.
2. Cohn ES, Kelley PM. Clinical phenotype and mutations in connexin 26 (DFNB1/GJB2), the most common cause of childhood hearing loss. Am J Med Genet 1999b;89:130-6.
3. Gasparini P et al. High carrie frequency of the 35delG deafness mutation in European population. Eur J Hum Genet 2000; 8:19023.
4. Green GE et al. Performance of coclear implant recipients with GJB2-related deafness. Am J Med Genet 2002;109:167-70.
5. Kenna MA, Wu BL, Cotanche DA, Korf BR, Rehm HL. Connexin 26 studies in patients with sensorineural hearing loss. Arch Otolaryngol Head Neck Surg 127:1037-42.
6. Liu Y, Ke X, Qi Li, Zhu P. Connexin 26 gene (GJB2): prevalence of mutations in Chinese population. N Hum Genet 2002;47(12):688-90.
7. Marazita ML et al. Genetic epidemiological studies of early-onset deafness in the US school-age population. Am J Med Genet 1993;46:486-91.
8. Morton NE. Genetic epidemiology of hearing impairment. Ann New York Acad Sci 1991;630:16-31.
9. Mustapha T, Arnos KS, Pandaya A. Advances in hereditary deafness. Lancet 2001;358:1082-90.
10. Oliveira CA, Maciel-Guerra AT, Sartorato EL. Deafness resulting from mutations in the GJB2 (connexin 26) gene in Brazilian patients. Clin Genet 2002;61:354-8.
11. Oliveira CA et al. Frequency of the 35delG mutation in the GJB2 gene in samples of European, Asian, and African Brazilians. Hum. Biol 2004;76: 313-6.
12. Pampanos A et al. Prevalence of GJb2 mutations in prelingual deafness in the Greek population. In J Pediatr Otorhinolaringol 2002;65:101-8.
13. Paparella MM, Fox RY, Schachern PA. Diagnosis and treatment of sensorioneural hearing loss in children. Otolaryngol Clin North Am 1989;22:51-74.
14. Sartorato EL et al. Determination on the frequency of 35delG allele in Brazilian noenatos. Clin Genet 2000;58(1):339-40.
15. Simões AM, Maciel-Guerra AT. A surdez evitável: predominância de fatores ambientais na etiologia da surdez neurossensorial profunda. J Ped 1992;68:254-7.
16. Simsek M et al. Absence of deafness associated connexin 26 (GJB2) gene mutations in the Omani population. Hum Mutat 18:545-6.
17. Sinnathuray AR, Toner JG, Clarke-Lyttle J, Geddis A, Patterson CC, Hughes AE. Connexin 26 (GJB2) gen-related deafness and speech intelligibility after cochlear implantation. Otol Neurotol 2004;25(4):935-42.
18. Sobe T et al. The prevalence and expression of inhered connexin 26 mutations associated with nonsyndromic hearing loss in the Israeli population. Hum Genet 2000;106:50-1.
Trabalho premiado no IV Congresso Triológico de 2005
1 Fonoaudióloga, Programa de Implantes Cocleares da Disciplina de Otorrinolaringologia e Cirurgia de Cabeça e Pescoço - UNICAMP.
2 Fonoaudióloga, Programa de Implantes Cocleares da Disciplina de Otorrinolaringologia e Cirurgia de Cabeça e Pescoço - UNICAMP.
3 Mestre, Laboratório de Genética Humana - CBMEG-UNICAMP.
4 Profa Dra; Laboratório de Genética Humana - CBMEG-UNICAMP.
5 Médico residente da Disciplina de Otorrinolaringologia e Cirurgia de Cabeça e Pescoço - UNICAMP.
6 Responsável pelo Programa de Implantes Cocleares da Disciplina de Otorrinolaringologia e Cirurgia de Cabeça e Pescoço - UNICAMP.
Endereço para correspondência: Paulo R. Cantanhede Porto - Alameda João Mendes Junior, 456 - Condomínio Vale do Itamaracá - Valinhos - SP 13270-000
Tel. (0xx19) 3233-6199.
Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) da RBORL em 10 de setembro de 2005.
Artigo aceito em 19 de setembro de 2005.
