INTRODUÇÃONefrite hereditária associada com perda auditiva sensório-neural (PASN) e anormalidades oculares é conhecida como Síndrome de Alport (SA)1.
Hurst, em 1923, descreveu a nefrite hereditária e Alport, em 19272, revendo a mesma família, salientou que a PASN era uma característica marcante em quase todos os membros1-3. Sohar (1956) identificou os defeitos oculares presentes nos doentes com SA1.
A SA é uma desordem hereditária caracterizada por hematúria, que leva freqüentemente à falência renal4. Pode ser acompanhada de manifestações extra-renais4. Muitas vezes, portanto, a nefropatia está associada com PASN e defeitos oculares4. Na Figura 1 está representada a complexa estrutura do colágeno tipo IV e a Figura 2 corresponde ao modelo tridimensional das cadeias a3.a4.a5 (hexâmero NC1)5.
O objetivo deste trabalho foi revisar os aspectos clínicos da perda auditiva na Síndrome de Alport e introduzir os recentes avanços genéticos e biomoleculares.
REVISÃO DE LITERATURAA freqüência estimada do gene responsável pela SA é de 1 em 5000, obtida de um amplo estudo, que observou 300 casos na população de 1,5 milhão habitantes de Utah6. Não está associada a qualquer raça ou região geográfica6.
A prevalência de indivíduos com SA, que necessitam de diálise/ transplante renal, é de 3,0% na Europa e de 2,2% nos EUA7.
GenéticaDuas formas são reconhecidas na base genética molecular da SA. A forma dominante ligada ao X, devido às mutações no lócus COL4A5 e a forma autossômica recessiva, resultando de mutações no lócus COL4A3 ou COL4A43. Entretanto, a análise genealógica sugere um tipo autossômico dominante. Em aproximadamente 85% dos casos de SA existe a forma dominante ligada ao X e em 15% dos casos existe a forma autossômica recessiva8.
Os seis genes colágeno tipo IV estão arranjados, em pares, em três cromossomos diferentes. As cadeias a1 e a2 humanas são codificadas pelos genes COL4A1 e COL4A2, pareados cabeça a cabeça, respectivamente, no cromossomo 13. Os genes COL4A3 e COL4A4 codificam as cadeias a3 e a4 do colágeno tipo IV, respectivamente e estão pareados cabeça a cabeça no cromossomo 2. As cadeias a5 e a6(IV) são codificadas, respectivamente, pelos genes COL4A5 e COL4A6, no braço longo do cromossomo X3.
Mutações no gene COL4A5 explicam possivelmente toda a SA ligada ao X9.
As cadeias a podem ser divididas em 3 domínios: o domínio 7S, amino-terminal (NH2, com aproximadamente 15 aminoácidos); o domínio central triplo-helicoidal (com aproximadamente 1400 aminoácidos) e a porção globular carboxi-terminal não colagenosa (NC1; COOH com perto de 230 aminoácidos)3. A seqüência principal é constituída por glicina (Gly), hidroxilisina (X) e hidroxiprolina (Y)10. (Figura 3)
Para o entendimento da SA é necessário que se conheça a estrutura do colágeno tipo IV. Existem seis cadeias a. As cadeias a1 e a2 são denominadas cadeias clássicas e as cadeias a3, a4, a5 e a6 são denominadas cadeias novas. As seis cadeias a, distintas geneticamente, estão arranjadas em três pró-colagenos triplo-helicoidais, que diferem na composição destas cadeias (Figura 4)5.
A formação da molécula precursora do colágeno tipo IV inicia-se pela interação do domínio NC1. A reunião de um trímero particular começa quando os 3 domínios NC1 iniciam uma interação molecular ainda desconhecida entre as 3 cadeias a. O próximo passo é a formação de um dímero colágeno tipo IV. Quatro moléculas precursoras interagem na região amino-terminal glicosilada 7S (NH2) para formarem tetrâmeros. Estas interações formam o núcleo da estrutura do colágeno tipo IV, que evolui para a supra-estrutura deste, com a ajuda de associações término-terminais e também laterais10.
As cadeias a1 e a2(IV) estão presentes em todas as membranas basais. As cadeias a3, a4 e a5(IV) expressam-se seletivamente nas membranas basais de alguns tecidos, incluindo os potencialmente acometidos na SA, que são: o rim (membrana basal glomerular/ MBG e membranas basais tubulares), a cóclea e o olho. As cadeias a5 e a6(IV) são características da pele, do músculo liso, do esôfago e do rim (cápsula de Bowman)3.
Uma mutação em uma das cadeias impede a incorporação das outras duas numa tríplice hélice estável11.
O colágeno tipo IV é o principal constituinte das membranas basais. As mutações presentes na SA produzem defeitos nas cadeias a3, a4 e a5(IV). O dano ao colágeno tipo IV, devido à mutação, rompe a função de ligação epitelial e leva ao defeito do órgão. Estes defeitos nas cadeias resultam no enlaçamento e na montagem incorreta dos monômeros, que serão degradados rapidamente. Estas mutações interrompem a substituição normal do desenvolvimento embrionário e causam a persistência das cadeias a1.a1.a2(IV) nas membranas basais renais, na cóclea e na cápsula do cristalino. A rede a1.a1.a2(IV) embriônica é mais susceptível à proteólise do que a a3.a4.a5(IV), pois esta última é mais fortemente ligada5.
Quadro Clínico e Critérios DiagnósticosEstes sinais são observados no quadro clínico da SA:
· Hematúria: a forma microscópica é a mais comum e a macroscópica vem em segundo lugar. Os homens apresentam hematúria microscópica persistente, com episódios de hematúria grosseira precipitada por infecções respiratórias altas12. A proteinúria é de menos de 1 a 2 g de proteína excretada por dia4,5.
· Falência renal progressiva4
· PASN (perda auditiva sensório-neural)4: intensidade variável, bilateral, simétrica e progressiva. Presente em aproximadamente 55% dos homens e 45% das mulheres7.
· Alterações oculares4: lentecone anterior (protrusão cônica ou esférica da superfície anterior do cristalino para a câmara anterior; conhecida como sinal da gota de óleo; ocorre em 10 a 30% dos casos; é patognomônico da SA)3,11, manchas oculares e catarata.
· Outros4: macro-trombocitopenia e leiomiomatose.
Os critérios diagnósticos que devem ser empregados são: história familiar de hematúria, com ou sem falência renal crônica; evidência eletro-microscópica de SA, na amostra de biópsia renal (espessura variável da MBG, com lâmina densa lamelada, circunscrevendo áreas claras contendo grânulos eletro-densos; presente em 60 a 90% dos casos); sinais oftalmológicos característicos descritos anteriormente e perda auditiva sensório-neural em tons agudos12.
Devem ser preenchidos 3 ou mais dos critérios estritos propostos acima12. Quanto ao curso clínico da SA: a progressão é mais rápida e previsível nos homens do que nas mulheres. Nas mulheres o curso clínico é mais variável, sendo possível uma vida normal e duradoura. Nos homens a hematúria é por toda a vida12.
Os meninos desenvolvem12:
· Hematúria na infância
· PASN progressiva durante os anos escolares
· Falência renal crônica
· Sinais oculares
O diagnóstico de SA deve basear-se: na história familiar e na investigação clínica de todos os membros da família, além do caso índice ou probando (membro afetado da família por meio do qual toda a família é avaliada)11. Estas informações constituem o padrão ouro para o diagnóstico de SA. A hematúria é a alteração mais freqüente8. A PASN nos tons agudos é um dos sinais mais úteis em um paciente com hematúria e sugere o diagnóstico até na ausência de uma biópsia renal ou de história familiar de doença renal12.
A perda auditiva é um dos primeiros sinais na SA1,7,12. Torna-se evidente no fim da infância ou no início da adolescência, em meninos com doença ligada ao X e além disto sua progressão sugere pobre prognóstico da doença renal13.
Analisando cinqüenta e um pacientes com SA, que exibiam perda auditiva, três configurações audiométricas foram identificadas: a forma escavada ocorreu em 47,1% dos casos, a descendente em 41,2% dos casos e a plana em 11,7% dos casos. A média dos limiares em 500, 1000 e 2000 Hz foi de 33dB NA na forma plana; 42 dB NA na forma escavada e de 50 dB NA na forma descendente. O padrão plano ocorreu por volta dos 8,5 anos de idade; o escavado aos 13,7 anos e o descendente aos 17,8 anos; o que poderia sugerir uma progressão de um padrão para o outro (Figura. 6)14.
A avaliação de 11 pacientes com SA, comprovada histologicamente, demonstrou limiares de recepção da fala compatíveis com as médias dos tons puros e índices de reconhecimento da fala em conformidade com a configuração das audiometrias. Observou-se recrutamento mais importante nas freqüências médias. A latência das ondas I, III e V e os intervalos I-V e III-V foram normais nesta descrição das respostas auditivas de tronco encefálico (ABR), confirmando a integridade das vias retro-cocleares e indicando que o defeito estava situado na cóclea1.
Outras nefrites hereditárias com perda auditiva devem ser afastadas, tais como: hiper-prolinemia, Doença de Charcot-Marie-Tooth e nefrite intersticial hereditária6.
Estudos em AnimaisForam possíveis as seguintes observações nos estudos em animais: as membranas basais (MBs) são o principal constituinte do labirinto membranoso e o componente mais comum das MBs é o colágeno tipo IV15,16.
As cadeias a3(IV), a4(IV), a5(IV) e a6(IV) foram localizadas, exclusivamente, nas membranas tectória e basilar, da cóclea de cobaias normais, por imuno-fluorescência indireta17. Estes resultados sugerem uma possível função das cadeias do colágeno tipo IV no ajuste ativo das membranas basilar e tectória, uma etapa essencial na discriminação da freqüência e na amplificação dos sinais auditivos17.
O exame eletro-microscópico, no rato mutante (Knockout; no qual um gene específico foi marcado, usado como modelo animal para a SA autossômica recessiva) revelou: adelgaçamento da membrana basal coclear e alteração na estria vascular, com edema nas células endoteliais e com redução do diâmetro interno dos capilares18. O adelgaçamento da membrana basal ao longo da membrana basilar pode ter um efeito na rigidez desta e a alteração na estria vascular poderia restringir o fluxo sanguíneo através deste tecido hiper-ativo metabolicamente18.
Estudos em cão Samoyed (modelo animal de SA ligada ao X) demonstraram a ausência das cadeias a3, a4 e a5 no ligamento espiral19. A perda destas cadeias nesta região poderia resultar numa capacidade reduzida dos miofibroblastos em manterem tensão suficiente na membrana basilar, com perda da percepção dos sons agudos. O sítio candidato para a alteração auditiva na SA humana seria o ligamento espiral19.
DISCUSSÃOA perda auditiva é um dos primeiros sinais e um dos mais úteis em um paciente com hematúria. A PASN nos tons agudos sugere o diagnóstico de SA até na ausência de uma biópsia renal ou de história familiar de doença renal; a biópsia pode ser arriscada nos casos de falência renal3,11 e o antecedente familiar é negativo nos casos isolados ou de mutação de novo8.
O otorrinolaringologista deve suspeitar desta afecção e deve incluir um exame de urina na investigação das perdas auditivas1. Crianças com hematúria inexplicada, adolescentes ou indivíduos de meia-idade do sexo masculino em EFFR e pacientes que apresentem história familiar de doença renal nos irmãos ou parentes do lado materno devem realizar um exame microscópico do sedimento urinário6. É importante solicitar a colaboração do geneticista para uma investigação quanto ao tipo de herança, para orientação quanto aos exames disponíveis de investigação genética e para o aconselhamento do paciente e da família. A contribuição do oftalmologista em pacientes com suspeita de SA é importante, pois o lentecone anterior é patognomônico e está consistentemente associado com uma rápida progressão para a falência renal e com a perda auditiva11.
Os métodos de avaliação auditiva (audiometria, ABR e otoemissões acústicas) devem ser empregados na investigação do caso índice e nos demais familiares (doentes e assintomáticos) para a pesquisa dos portadores. O diagnóstico dos portadores de doenças genéticas é extremamente valioso para o aconselhamento dos pacientes e das famílias20.
O acompanhamento cuidadoso da perda auditiva é um fator relevante para o prognóstico da evolução da doença renal. Homens com perda auditiva nas duas primeiras décadas de vida tinham doença renal mais severa13.
O acúmulo de metabólitos poderia ter um efeito ototóxico21. No entanto, existem vários relatos de PASN precedendo ou ocorrendo na ausência de alterações renais7. As similaridades entre o rim e a cóclea, com respeito à regulação do desenvolvimento pós-natal das cadeias do colágeno tipo IV, sugerem que a alteração otológica na SA deve ser decorrente de um defeito nas MBs cocleares e não por um comprometimento renal5,16.
No campo da investigação auditiva, muitos avanços recentes ocorreram pelo uso de cobaias e de modelos animais para a perda auditiva da SA humana. Permitiram a compreensão da audição normal a nível molecular e os mecanismos que são alterados quando uma determinada mutação ocorre22. As investigações no rato Knockout COL4A3 sugeriram alterações na estria vascular como causa da perda auditiva na SA18 e no cão Samoyed o ligamento espiral na orelha interna pode ser responsável pela PASN nos sons agudos19. Os estudos em orelhas humanas de pacientes com SA são prejudicados devido a dificuldades de obtenção de ossos temporais, o limitado número de peças para estudo, as dificuldades na fixação e o processo de autólise após a morte, contribuindo para as diferenças observadas nas descrições destes ossos temporais na literatura16.
Tratamento e PerspectivasA avaliação regular da audição de pacientes com SA é importante. As anormalidades oculares nestes doentes prejudicam as pistas visuais, essenciais para a comunicação de pacientes com perdas auditivas severas. A reabilitação cuidadosa da PA é fundamental1.
Em relação ao tratamento da SA, a diálise e o transplante renal são indicados neste grupo de pacientes, com SA e estágio final de falência renal (EFFR). Durante a hemodiálise ocorrem alterações osmóticas e eletrolíticas na endolinfa. Pacientes pós-transplante renal usam medicamentos imunossupressores (ciclosporina A e corticosteróides), que causam alteração na viscosidade do plasma e na circulação da orelha interna. Estes pacientes necessitam, portanto, de acompanhamento otológico1,3,6,11,23.
O prognóstico melhorou com o transplante renal, aumentando a longevidade dos pacientes com SA e falência renal. Portanto, o acompanhamento, a reabilitação e a investigação da evolução da perda auditiva tornam-se agora importantes na qualidade de vida deles1.
Alguns autores relataram melhora ou estabilização da perda auditiva em pacientes pós-transplante renal1,21, mas outros observaram piora23. Com a melhora da função renal pós-transplante é mais fácil compreender que ocorra uma estabilização ou até mesmo uma progressão mais lenta da perda auditiva, já que esta decorre da alteração das membranas basais, pelo dano ao colágeno tipo IV. Serão necessários estudos adicionais para esclarecer a evolução da alteração auditiva neste grupo de pacientes pós-transplante renal.
É possível construir um perfil dos pacientes que podem desenvolver nefrite anti-MBG, pós-transplante renal: geralmente homens, sempre com perda auditiva e que desenvolveram EFFR antes dos 30 anos3. A nefrite anti-MBG, pós-transplante renal é rara, porém grave, sendo de interesse prever a ocorrência desta. A avaliação da audição pode contribuir para a elaboração deste perfil de risco.
A terapia gênica está sendo pesquisada em cães Samoyed (modelo animal para a SA ligada ao X). O emprego de um vetor adenoviral, codificando a cadeia a5(IV) do colágeno, nas células do músculo liso da bexiga destes animais, corrige o defeito e restaura a expressão das cadeias a5 e a624. Questiona-se ainda em que fase da doença renal será adequada esta modalidade de tratamento3. Para manter a expressão destas cadeias, até então desconhecidas pelo hospedeiro, necessita-se de imunossupressão e discute-se qual o melhor vetor3. Os estudos relativos à terapia gênica coclear contribuem para a análise genética molecular da audição e experimentalmente a habilidade para introduzir genes na orelha interna pode levar a elucidação da função de proteínas cocleares e ao controle de genes específicos da orelha interna25. Os métodos de introdução do vetor incluem infusão por mini-sondagem ou micro-injeção na rampa timpânica, via janela redonda25.
É necessário tecer alguns comentários sobre outras desordens. A Síndrome unha-rótula decorre de uma mutação autossômica dominante no fator de transcrição LMX1B, que regula os genes COL4A3 e COL4A4 e as crianças têm síndrome nefrótica e displasias esqueléticas e das unhas5. A Síndrome de Goodpasture é uma desordem imune, com anticorpos séricos dirigidos contra regiões específicas da cadeia a3(IV) do colágeno tipo IV e os pacientes apresentam hemorragia pulmonar com nefrite, é fatal se não tratada; portanto a patogênese entre a SA e a Síndrome de Goodpasture está ligada ao mesmo pró-colágeno a3.a4. a5(IV)5. O paciente com diabetes mellitus não-controlado desenvolve glomerulonefrite progressiva associada com PASN e com doença degenerativa da retina; já foi confirmado, por imuno-fluorescência, que no rim e na retina destes pacientes, o RNAm COL4A1 e o RNAm COL4A2 estão elevados, pela ativação de uma via secundária por metabólitos glicosilados15. Trabalhos nesta área irão fornecer um novo conhecimento a respeito das doenças relacionadas ao colágeno.
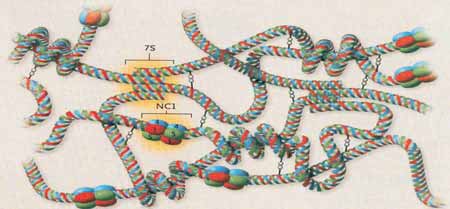
Figura 1. Montagem e organização da rede de colágeno tipo IV (Hudson et al., 2003).
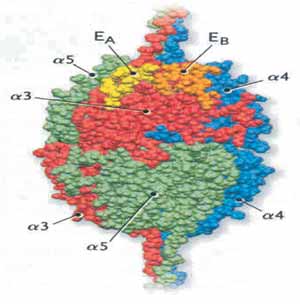
Figura 2. Modelo tridimensional do hexâmero NC1 a3.a4.a5(IV). Em vermelho a3, em azul a4 e em verde a5. As localizações EA (amarelo) e EB (dourado) representam os domínios para os auto-anticorpos da Síndrome de Goodpasture (Hudson et al., 2003).
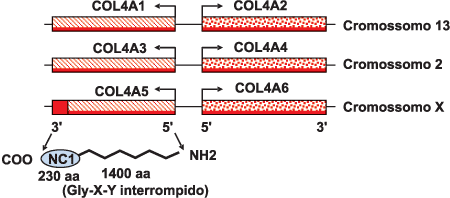
Figura 3. Organização genômica dos seis genes colágeno tipo IV, que estão distribuídos nos cromossomos 13, 2 e X. Os 5 éxons na porção 3' terminal do COL4A5, codificam o domínio NC1 do a5(IV) e estão em vermelho (Kashtan & Michael, 1996).

Figura 4. Organização triplo-helicoidal da família do colágeno tipo IV. A seleção das cadeias para associação em uma molécula precursora é governada pelo reconhecimento dos domínios NC1 (Hudson et al., 2003).
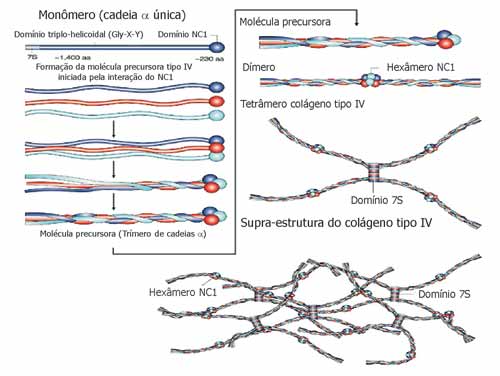
Figura 5. Formação da rede colágeno tipo IV (Kalluri, 2003).
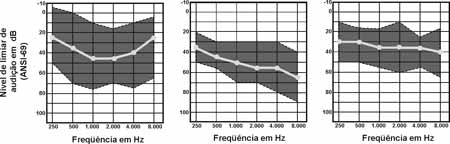
Figura 6. Classificação de 51 audiometrias pelo tipo de configuração. (A) escavado, (B) descendente e (C) plano (Rintelmann, 1976).
Após a análise crítica da literatura as seguintes conclusões foram possíveis:
1. A SA caracteriza-se por hematúria, que evolui para falência renal e pode ser acompanhada de manifestações extra-renais. A perda auditiva é um achado extra-renal freqüente e um dos primeiros sintomas na SA, sendo um fator relevante para o prognóstico da evolução da doença renal.
2. A SA é genética e decorre da alteração das cadeias do colágeno tipo IV nas membranas basais.
3. A perda auditiva é SN, de intensidade variável, progressiva e simétrica. Acomete as freqüências médias e altas.
4. Na investigação das PASN nas crianças com hematúria, nos adolescentes e adultos masculinos em EFFR, nos pacientes com história familiar de doença renal em irmãos ou parentes do lado materno, deve-se incluir um exame de urina.
5. O otologista deve atuar no acompanhamento destes pacientes. Deve solicitar uma avaliação do oftalmologista e do geneticista para o aconselhamento dos pacientes e da família.
REFERÊNCIAS BIBLIOGRÁFICAS1. Gleeson MJ. Alport's syndrome: audiological manifestations and implications. J Laryngol Otol 1984; 98(5): 449-65.
2. Alport AC. Hereditary familial congenital hemorrhagic nephritis. Br Med J 1927; 1: 504-6.
3. Kashtan CE, Michael AF. Alport syndrome. Kidney Int 1996; 50(5): 1445-63.
4. Barker DF, Pruchno CJ, Jiang X, Atkin CL, Stone EM, Denison JC, et al. A mutation causing Alport syndrome with tardive hearing loss is common in the Western United States. Am J Hum Genet 1996; 58(6): 1157-65.
5. Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Mechanisms of the disease: Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Engl J Med 2003; 348: 2543-56.
6. Atkin CL, Gregory MC, Border WA. Alport syndrome. In: Schrier RW, Gottschalk CW. Diseases of the kidney. 4thed., Boston: Little, Brown, 1988. p.617-41.
7. Wester DC, Atkin CL, Gregory MC. Alport syndrome: clinical update. J Am Acad Audiol 1995; 6(1): 73-9.
8. Gross O, Netzer KO, Lambrecht R, Seibold S, Weber M. Meta-analysis of genotype-phenotype correlation in X- linked Alport syndrome: impact on clinical counseling. Nephrol Dial Transplant 2002; 17(7): 1218-27.
9. Barker DF, Hostikka SL, Zhou J, Chow LT, Oliphant AR, Gerken SC, et al. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 1990; 248: 1224-7.
10. Kalluri R. Basement membranes: structure, assembly and role in tumor angiogenesis. Nat Rev Cancer 2003; 3(6): 422-33.
11. Pirson Y. Making the diagnosis of Alport's syndrome. Kidney Int 1999; 56(2): 760-75.
12. Flinter FA, Cameron JS, Chantler C, Houston I, Bobrow M. Genetics of classic Alport's syndrome. Lancet 1988; 2: 1005-7.
13. Cassady G, Brown K, Cohen M, DeMaria W. Hereditary renal dysfunction and deafness. Pediatrics 1965; 35: 967-79.
14. Rintelmann FW. Auditory manifestations of Alport's disease syndrome. Tr Am Acad Ophth Otol 1976; 82: 375-87.
15. Cosgrove D, Samuelson G, Pinnt J. Immunohistochemical localization of basement membrane collagens and associated proteins in the murine cochlea. Hear Res 1996; 97: 54-65.
16. Cosgrove D, Kornak JM, Samuelson G. Expression of basement membrane type IV collagen chains during postnatal development in the murine cochlea. Hear Res 1996; 100: 21-32.
17. Kalluri R, Gattone IIª VH, Hudson BG. Identification and localization of type IV collagen chains in the inner ear cochlea. Connect Tissue Res 1998; 37: 143-50.
18. Cosgrove D, Samuelson G, Meehan DT, Miller C, McGee J, Walsh EJ, et al. Ultrastructural, physiological, and molecular defects in the inner ear of a gene-knockout mouse model for autosomal Alport syndrome. Hear Res 1998; 121: 84-98.
19. Harvey SJ, Mount R, Sado Y, Naito I, Ninomiya Y, Harrison R, et al. The inner ear of dogs with X-linked nephritis provides clues to the pathogenesis of hearing loss in X-linked Alport syndrome. Am J Pathol 2001; 159(3): 1097-104.
20. Sirimanna KS, France E, Stephens, SDG. Alport's syndrome: can carriers be identified by audiometry? Clin Otolaryngol 1995; 20: 158-63.
21. McDonald TJ, Zincke H, Anderson CF, Ott NT. Reversal of deafness after renal transplantation in Alport's Syndrome. Laryngoscope 1978; 88: 38-42.
22. Avraham KB. Mouse models for deafness: lessons for the human inner ear and hearing loss. Ear Hear 2003; 24(4): 332-41.
23. Sefer S, Trotic R, Lacmanovic V, Degoricia V, Ratkovic-Gusic I, Kes P. Effects of renal transplantation on hearing and ocular changes in a monozygotic twin with Alport's syndrome: comparison with other twin on hemodialysis. Croat Med J 2000; 41(2): 203-6.
24. Harvey SJ, Zheng K, Jefferson B, Moak P, Sado Y, Naito I, et al. Transfer of the a5(IV) collagen chain gene to smooth muscle restores in vivo expression of the a6(IV) collagen chain in a canine model of Alport syndrome. Am J Pathol 2003; 162(3): 873-85.
25. Lalwani AK, Mhatre AN. Cochlear gene therapy. Ear Hear 2003; 24(4): 342-8.
1 Mestranda em Otorrinolaringologia pela Faculdade de Ciências Médicas da Santa Casa de São Paulo, Médica Preceptora da
Clínica de Otorrinolaringologia do Hospital do Servidor Público Municipal.
2 Professor Adjunto do Departamento de Otorrinolaringologia da Faculdade de Ciências Médicas da Santa Casa de São Paulo.
Trabalho realizado no Departamento de Otorrinolaringologia da Santa Casa de São Paulo.
Endereço para correspondência: Dra. Fátima R. A. Alves - Av. Moema, 801 São Paulo SP 04077-023.
Tel.: (0xx11) 5052-4344 - Fax: (0xx11) 5052-4336 - E-mail: ffraa@aol.com
Artigo recebido em 09 de março de 2004. Artigo aceito em 11 de maio de 2004.
