IntroduçãoO Carcinoma de Células Escamosas Oral (CCEO) que também recebe o nome de Carcinoma Epidermóide ou Espinocelular representa mais de 90% de todos os tumores malignos que afetam a cavidade bucal.1 Acomete, principalmente, o sexo masculino na faixa etária dos 50 aos 80 anos de idade.1 O uso do tabaco e o consumo de álcool são fatores de risco bem estabelecidos na maioria dos casos.1 Entretanto, uma pequena proporção (15-20%) ocorre em pacientes sem história de tabagismo e etilismo, sugerindo a presença de outros fatores de risco.2
Uma importante constatação, proveniente de estudos epidemiológicos da doença, mostra aumento do desenvolvimento de CCEO em pacientes menores de 45 anos de idade, a partir da década de 80.3,4 O comportamento desse tumor em adultos jovens é controvertido, havendo relatos de maior anaplasia e pior prognóstico, em comparação a habitual faixa etária.1,3-5 Adicionalmente, a implicação de agentes com conhecido potencial carcinogênico como o tabaco e o álcool, nesse grupo, é pouco definida e comumente não relatada.1,3-5 Desse modo, outros fatores vêem sendo investigados, principalmente de natureza microbiana e molecular.1,3-5
Uma comparação de casos de CCEO em pacientes com menos de 40 anos de idade entre as décadas de 60 e 70 e os dias de hoje mostra que a incidência de CCEO em adultos jovens dobrou, chegando a cerca de 6% do total de casos de CCEO.4 Tal fato pode ser considerado coincidente com uma maior preocupação atual com os levantamentos epidemiológicos e os relatos de caso. Entretanto, comparações entre as décadas de 80 e 90 demonstram a mesma elevação da taxa.3
Nos Estados Unidos, as taxas de mortalidade por CCEO vêm diminuindo em homens brancos de idade elevada, possivelmente em correlação com a redução do uso do tabaco na população.3 Em contrapartida, a taxa de mortalidade por CCEO em língua de pacientes jovens do sexo masculino vem crescendo, demonstrando pouca resposta ao tratamento.3
Uma das principais causas que levam à limitação da resposta ao tratamento, prognóstico sombrio e diminuição da sobrevida em pacientes portadores de câncer é o atraso no diagnóstico da lesão.10 Nos casos de CCEO, essa demora reflete-se nos processos movidos contra profissionais de saúde nos EUA.10 As principais queixas citadas são o manejo inadequado do paciente, negligência no diagnóstico, não realização de procedimentos com fins diagnósticos (biópsia), demora no encaminhamento ao profissional competente e as complicações cirúrgicas.10 A queixa mais comum - negligência no diagnóstico - responde por mais de 86% das causas ganhas pelos pacientes, quando a demora é igual ou maior a 3 meses.10 Existe uma certa dificuldade na definição do termo "demora no diagnóstico", sendo que a maioria dos trabalhos e processos movidos admite um período mínimo de 3 meses. 10 No entanto, este tempo tende a ser maior em pacientes jovens, uma vez que os profissionais com menos experiência não prevêem a possibilidade de malignidade e tendem a tratar a lesão como se esta fosse benigna.10
Segundo o estudo de Lydiatt (2002), 45% dos processos de pacientes com CCEO são movidos contra cirurgiões dentistas.10 Entre estes profissionais, 60% relatam que devem, mas não realizam a biópsia.10 De acordo com o autor, tal fato ocorre porque os dentistas clínicos gerais não se sentem confortáveis para realizar tal procedimento.10 Os pacientes alegam que os otorrinolaringologistas também falham no diagnóstico precoce do câncer, mas esses profissionais realizam biópsia e fazem o referenciamento do paciente mais freqüentemente.10 No entanto, devido à prática odontológica estar menos relacionada a condições malignas, os dentistas conseguem defesa em um maior número de processos, quando comparados aos otorrinolaringologistas.10
Ademais, vale ressaltar que a média de idade dos pacientes que procuram auxílio jurídico é de 45 anos, ou seja, jovens para o CCEO.10 Apesar da implicação do tabaco e álcool como importantes fatores de risco para o CCEO, estes são reportados somente por um pequeno número de pacientes jovens como dito anteriormente, mas mesmo nos casos onde há correlação, alega-se que nesse grupo a exposição aos carcinógenos seria de curta duração e insuficiente para o desenvolvimento de uma lesão maligna.3-5,10,11Além disto, um enorme número de pessoas são expostas a tais fatores de risco e somente uma pequena proporção destes desenvolve CCEO.12 Isso tem levado à procura de outros fatores de risco como deficiências imunológicas, nutricionais, fatores genéticos e participação de agentes microbiológicos. Dentre esses vem sendo considerada a contribuição de vírus com conhecido potencial oncogênico, como o Papilomavírus humano (HPV) e o vírus Epstein-Barr (EBV) que há cerca de 20 anos vêm sendo associados ao CCEO.8,13
Levando em conta a necessidade de maiores esclarecimentos acerca das características biológicas, e principalmente etiológicas do CCEO, em adultos e adultos jovens, propomos uma análise das informações referentes a fatores etiológicos comumente associados ao câncer de boca como o Papilomavírus humano (HPV), o vírus Epstein-Barr (EBV) e a proteína p53. Avaliamos ainda a participação da telomerase, uma das enzimas que auxiliam na manutenção da capacidade replicativa da célula.6-8 O aumento da expressão da telomerase tem sido descrito no CCEO e em tumores malignos de outros sítios como o útero, pulmão, fígado, mama, entre outros.6,9
Revisão de literaturaPapel Viral
A etiologia multifatorial do câncer é amplamente aceita, entretanto, além do papel dos fatores de risco ambientais e/ou fatores genéticos, uma etiologia infecciosa vem sendo defendida.14 Inicialmente, a sífilis foi implicada na etiologia do CCEO e subseqüentemente, espécies de Cândida foram incriminadas, entretanto, recentemente o interesse tem se voltado para a investigação da etiologia viral no CCEO.1
O equilíbrio entre a proliferação e a morte celular (apoptose) é de vital importância para a sobrevivência de qualquer organismo vivo, e este equilíbrio pode ser quebrado por uma infecção viral, que pode culminar em transformação neoplásica.14 Numerosos produtos genéticos de origem viral podem ligar-se a genes regulatórios da proliferação e morte celular e alterar suas funções.14 O sinal inicial da apoptose pode ser inibido por proteínas de adenovírus (E1B 19 kDa e E3), mixomavírus (MT2), bacilovírus (iap), herpes simples vírus e citomegalovírus, e induzido por mixovírus e vírus da hepatite C.14 Além disso, a transdução de sinais para a morte celular (p53, pRb, bcl-2) e a transativação de protooncogenes (c-myc, c-fos, c-jun) pode ser inibida por proteínas de adenovírus, EBV, citomegalovírus, entre outros.14 Como conseqüência, as células hospedeiras com infecção viral latente são levadas à proliferação sem regulação por reparo celular ou mesmo mecanismos de eliminação de células lesadas.14 Essas células podem continuar acumulando mutações induzidas por outros mecanismos capazes de causar dano ao DNA, como o tabaco, o álcool, carcinógenos, toxinas, entre outros, o que pode resultar no desenvolvimento de uma neoplasia maligna.14
Papilomavírus Humano (HPV)
O HPV é um vírus DNA pequeno, não-envelopado da família papovaviridae, com tropismo por células epiteliais que pode induzir hiperplasia, papilomatose e lesões verrucosas no epitélio estratificado escamoso de pele e mucosas.8,15 Já foram identificados mais de 100 diferentes tipos de HPV em humanos.,8,15,16 Alguns destes, como o HPV 16, 18, 33 e 58, parecem ter um importante papel no desenvolvimento de certos tumores humanos, sendo considerados de alto risco.8,12,15,17
A infecção por HPV representa um fator de risco extremamente significativo para o Carcinoma de cérvix uterino.8,12,13,15-18 Os HPV de alto risco são a maior causa de cânceres anogenitais e têm sido implicados no câncer de cabeça e pescoço, pois a presença de DNA viral tem sido identificada nestes tumores.8,12,15,17 No entanto, seu papel no CCEO não foi ainda bem definido.12,15-18
A infecção por HPV é comumente identificada pela detecção de DNA viral em células e tecidos, no entanto, como a infecção por HPV é focal ocorrem erros freqüentes, como falsos positivos e falsos negativos, principalmente em indivíduos assintomáticos.17 No entanto, como a infecção por HPV é transitória, a ausência de DNA do HPV não descarta uma infecção prévia.17 Anticorpos para os antígenos do capsídio do HPV demonstram ser marcadores confiáveis de infecção anterior e/ou presente e são usados em pesquisas séricas, onde a detecção costuma ser mais confiável.8,17
Desde 1977 mais de 600 casos de carcinoma oral têm sido estudados pela identificação de HPV.18 A prevalência citada, por diferentes técnicas, varia de 6% a 94%, sendo que o método de maior sensibilidade para o HPV parece ser a reação por cadeia polimerase (PCR)8,15,18,19 Apesar disto, a média de prevalência é menor que no câncer cervical.12,18 Entretanto, assim como no trato genital, os HPV-16 e 18 são de longe os tipos mais comumente encontrados, representando 80% das lesões HPV positivas.12,17,18
O HPV de alto risco é capaz de imortalizar células epiteliais orais e cervicais in vitro.17 As oncoproteínas E6 e E7 são os dois maiores oncogenes virais expressos no tecido neoplásico e ambos estimulam a proliferação celular, sendo capazes de se ligar e inativar proteínas supressoras tumorais do hospedeiro, como a p53 e pRb, respectivamente.20A E6 pode ainda ativar telomerase, outro fator envolvido na carcinogênese.8,16 Assim sendo, as oncoproteínas virais são capazes de transformar ceratinócitos primários humanos dos tratos genital e respiratório superior, desequilibrando o mecanismo regulador do ciclo celular, levando a uma progressão genética para CCEO.8,17 Conseqüentemente, a infecção por HPV pode representar uma alternativa, funcionalmente comparável, para os mecanismos moleculares da carcinogênese.2
Observa-se ainda uma tendência maior à positividade para HPV em pacientes não-etilistas e não-tabagistas, quando comparados aos usuários de tais substâncias.2 No entanto, a maior parte dos estudos concorda que não há correlação estatística entre a prevalência de HPV e a história de consumo de tabaco, embora ambas participem da carcinogênese na mucosa oral.15
Um fato inesperado relatado é a redução de aproximadamente 40% no risco de óbito de pacientes com tumores HPV positivos.2 Um fato inesperado, pois estes tumores foram menos associados ao álcool e tabaco, e a estes não se pode aplicar o conceito de cancerização de campo, pois as infecções por HPV tendem a ser focais, o que primariamente deveria levar a um pior prognóstico, o que não é relatado.2 Os carcinomas HPV positivos parecem ser uma entidade distinta (acometem mais células basais e tem um componente inflamatório menor), com biologia distinta (menor mutação de p53), fatores de risco distintos (menor associação com tabaco e álcool) e um curso clínico distinto (maior sobrevida).2 A maior prevalência de positividade em CCEO é para o HPV de tipo 16, que chega a ser duas vezes maior do que em grupos controles, não havendo diferença de positividade entre homens e mulheres.2 Observa-se ainda maior positividade para HPV em adultos jovens e este fato seria responsável por uma diferença biológica, que neste caso aponta para um melhor prognóstico, fato contrário ao esperado.16
Um estudo demonstra a correlação entre a detecção por PCR de HPV sérico e lesional e constata que os pacientes que apresentam HPV lesional apresentam HPV sérico somente nos estágios mais avançados da doença, sendo que quatro destes seis pacientes apresentaram metástase à distância e 50% destes pacientes com HPV sérico foram a óbito.19 No entanto, os 86% pacientes que apresentaram HPV lesional e ausência de HPV sérico estão sem evidência da doença.19 Assim sendo, a presença de HPV sérico estaria correlacionada a um estágio mais avançado da doença e de pior prognóstico.19
Parece haver uma relação entre a infecção local de HPV e a sua detecção em outros sítios.18 O HPV tem o potencial de invadir outras células epiteliais após a infecção primária de um sítio epitelial.18 Premoli-De-Percoco et al. (1998), utilizando hibridização in situ em mulheres com CCEO, demonstrou a presença simultânea de HPV na citologia cervicovaginal e no CCEO em 23 de 28 casos.18 Neste caso, a positividade para HPV em CCEO foi maior na faixa etária dos 50-69 anos, grupo em que se esperaria uma menor prevalência de HPV.18 A história natural da infecção por HPV demonstra um pico entre as mulheres sexualmente ativas (15-25 anos) e tende a estabilizar-se após os 30 anos, devido à resposta imunológica, quando as mulheres geralmente apresentam resolução da infecção. No entanto, outras sofrem uma pequena displasia epitelial que depois de um longo período de incubação, combinado com fatores ambientais, culmina na transformação maligna das células epiteliais.18
Esptein-Barr vírus (EBV)
O EBV, membro do grupo dos vírus herpes humanos, apresenta DNA de cadeia dupla e tem a habilidade de produzir uma infecção latente caracterizada por uma baixa expressão de genes virais e mínimos efeitos citopáticos ou de proliferação viral.21,22 O EBV apresenta alta prevalência na população, de forma que cerca de 90% dos adultos demonstram anticorpos para EBV e costuma infectar linfócitos e células epiteliais.21,22 O EBV é bem estabelecido como agente etiológico do linfoma de Burkitt e do carcinoma nasofaríngeo.21,23,24
O EBV normalmente não replica nos linfócitos B recém-infectados.25 Nestes linfócitos B infectados, porém latentes, existe a expressão de seis diferentes proteínas nucleares ou EBNAs, duas diferentes proteínas de membrana ou LMP e dois pequenos filamentos de RNA ou EBERs.25 Estes produtos virais mantêm a latência e estimulam o linfócito em repouso a se proliferar continuamente.25 Apesar de latência significar ausência de infecção lítica, este termo se torna confuso e inadequado para o EBV, uma vez que na infecção por EBV na infecção latente ocorrem crescimento e proliferação celular imediatos e eficientes.25 No entanto, apesar da presença de genomas e produtos virais, a proliferação do linfócito B infectado é similar àquela ocorrida pela ação de um antígeno, mitógeno ou Il-4.25 De qualquer forma, na latência do EBV ocorre a proliferação da célula infectada, e não de partículas virais como ocorre na infecção lítica viral.25
A latência pode ser diretamente induzida para o ciclo lítico pela a ativação da BZLF (zebra), uma proteína transativadora do EBV.25 (Figura 1) O ciclo lítico é caracterizado por uma intensa transcrição, replicação de DNA e pela produção de proteínas tardias como antígenos de capsídeos e glicoproteínas.25A Leucoplasia Pilosa Oral é a doença causada por replicação focal, sendo a única manifestação in vivo da infecção replicativa do EBV.25
A proteína LMP-1 modula o crescimento e a diferenciação, induz a expressão de múltiplos marcadores de superfície celular, ativadores celulares, antígenos e moléculas de adesão.23,24 A LMP-1 reduz a resposta celular à sinais de diferenciação, aumenta a invasividade da mesma na matriz colágena e pode transformar fibroblastos humanos em ceratinócitos.22-24 Pode ainda induzir resistência à apoptose através da ativação de proteínas anti-apoptóticas como a Bcl-2.23,24 Estudos in vitro demonstram que a LMP-1 pode bloquear a apoptose mediada por p53 em células epiteliais e em células de linfoma de Burkitt.23,24 A expressão de LMP-1 em indivíduos imunodeprimidos pode induzir a transformação de linfócitos B e a aparição de processos linfoproliferativos.24
Além disso, vem sendo associado a uma série de neoplasias malignas em geral, como o carcinoma tímico, o carcinoma gástrico, o câncer de mama e o CCEO.21-23 A ação oncogênica do EBV parece envolver a oncoproteína latente de membrana 1 (LMP-1).22-24
Uma alta prevalência de EBV foi demonstrada em CCEO.22 No entanto, a prevalência dessa associação varia muito de acordo com a região geográfica e as técnicas empregadas.21,22 Utilizando a PCR, técnica mais sensível, a prevalência giraria em torno de 37,9%, sem diferença estatística entre fumantes e não-fumantes, etilistas e não-etilistas.21
Um estudo para avaliar o DNA, RNA e nível de proteínas do EBV no CCEO demonstrou uma alta prevalência por PCR de DNA de EBV(100%).22 No entanto, não demonstrou a presença por imuno-histoquímica de LMP-1 nem de BZLF-1(Zebra), o que sugere que o EBV não apresenta potencial transformador, devido a ausência de LMP-1; e nem ciclo lítico, por ausência de Zebra nos casos de CCEO.22 Tal achado sugere que o EBV não apresenta potencial carcinogênico no CCEO.22
Alterações Genéticas - P53
Um outro fator comumente associado à etiologia do CCEO é a mutação da p53.20,26-28 Desde a descoberta da p53, no final da década de 70, acredita-se que esta seja responsável pela modulação da resposta celular à um estresse endógeno ou exógeno, como danos ao DNA, hipóxia, ativação de oncogenes, replicação viral e depleção de ribonucleotídeos, sendo um importante gene de supressão tumoral.20,27,30
A p53 pode bloquear o ciclo celular na fase G1 de células com danos subletais em seu genoma até o completo reparo deste.27 Além disto, pode induzir apoptose, evitando o desenvolvimento de clones de células com graves danos ao DNA.27A ausência da p53 nativa, seja por inativação ou destruição, e algumas mutações da proteína nativa são capazes de impossibilitar a função de guardiã do genoma exercida pela p53, permitindo assim a sobrevida de células com danos em seu DNA (Figura 2).20,27,28
A associação entre o consumo de tabaco e álcool e a mutação da p53 é controvertida.26 Entretanto, estudos genéticos demonstram que a região mutada do gene é diferente: em pacientes com fatores de risco como tabaco e álcool, pacientes sem fatores de risco e pacientes com carcinoma de lábio.20,26 Tal diferença mutacional, nestes casos, sugere etiologias distintas, tais como estímulos exógenos, endógenos e ação dos raios ultravioletas, respectivamente.20,26
Uma alta freqüência de CCEO ocorre na ausência de mutação da p53, onde parece existir apenas uma inativação da função, seja por destruição da proteína ou inativação da mesma, como na infecção por HPV que causa proteólise da p53, ou devido a uma superexpressão de MDM2 que leva a uma inativação da p53.20,28 Todas estas alterações na p53 permitem que a célula escape do mecanismo de controle da proliferação, desempenhando um importante papel na imortalização celular.28
Telômeros e Telomerase
Os telômeros são estruturas especializadas existentes na porção terminal dos cromossomos das células eucarióticas, compostos por repetições da seqüência TTAGG.6,7,29 Os telômeros protegem os cromossomos de quebra enzimática do DNA, replicação incompleta, previnem recombinações e fusões aberrantes, e asseguram a completa replicação do cromossomo durante a divisão celular.7,9,29 Os telômeros humanos, de células somáticas, sofrem um encurtamento progressivo durante a divisão celular, onde há uma perda da seqüência terminal do DNA decorrente da replicação.6,7,9,29 A perda progressiva do telômero causa senescência.6,7,9,29
Quando há grande encurtamento do telômero, a célula pára de se dividir.6,7 Assim sendo, o telômero é reconhecido como o "relógio mitótico", sendo responsável pela capacidade replicativa celular.6 Entretanto, células germinativas e células tronco escapam do encurtamento do telômero por expressar telomerase, uma enzima que sintetiza DNA telomérico.7
A telomerase, uma DNA polimerase ribonucleoproteína, contém um componente RNA que sintetiza a repetição DNA do telômero, compensando a perda dos telômeros durante a divisão celular.29 Teoricamente, acredita-se que somente células germinativas possuam atividade da telomerase, mas, recentemente, alguns tipos de células como as da medula óssea, de tecido gastrintestinal e da camada basal da pele apresentaram, embora fraca, atividade da telomerase.7
A ausência de telomerase em células normais resulta em erosão progressiva do telômero, levando a uma replicação incompleta, que gera instabilidade cromossomial, causando senescência6 Nesse último caso, a senilidade celular ocorre porque os telômeros estão encurtando.7 Telômeros curtos demais impedem a célula de se replicar, mas também causam instabilidade cromossômica e, portanto, tendência a mutações.6 Desta forma, a telomerase agiria como um fator que potencializaria o crescimento tumoral.30
A P53, por sua vez interpreta o encurtamento do telômero como "erros de DNA" e impede a célula de continuar a replicar levando-a a um estado de senilidade.6 E como, normalmente, não há telomerase na célula, ela pára de se dividir.6 O estado senil da célula é considerado um mecanismo de proteção anti tumoral (Figura 3).6,28
A telomerase tem sido expressa em alta porcentagem em extratos de vários tipos de câncer.6,7,28 Apresenta expressão em 75% dos CCEO, 80% dos cânceres de pulmão, 84% dos cânceres de próstata, 85% em fígado, 93% dos cânceres de mama, entre outros.9 Linhagens de células imortalizadas, tanto espontaneamente quanto dependente de transformação por oncovírus como o HPV, geralmente são telomerase positivas.9 A oncoproteína E6 dos HPV de alto risco é capaz de ativar a telomerase, por mecanismos ainda desconhecidos.6,9,28 A atividade da telomerase tem sido detectada também em tecidos normais, indicando que a telomerase pode ser um marcador de proliferação e não somente de carcinogênese.7,9,29
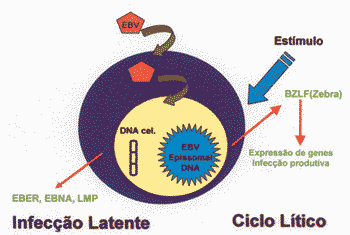
Figura 1. Produtos da infecção por EBV na célula humana, produção de EBER, EBNA e LMPs na infecção latente e expressão de BZLF-1 (Zebra) e LMP-1 no ciclo lítico.
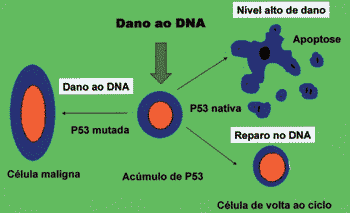
Figura 2. Função do p53 no controle da proliferação. A P53 mutada ou inativa leva à transformação maligna.
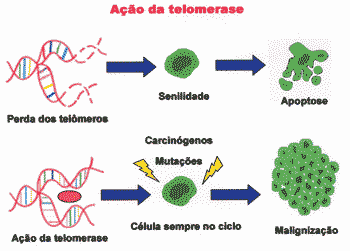
Figura 3. A progressiva perda dos telômeros leva à senilidade, enquanto a ação da telomerase leva a uma contínua multiplicação, o que pode levar a malignidade.
Relatos do envolvimento do HPV na carcinogênese oral são conflituosos, com taxas de infecção que variam de 0 a 87%.15 Os resultados encontrados na literatura, por sua vez, são de difícil interpretação devido ao tamanho das amostras, método de coleta do tecido, conservação do tecido, sensibilidade do método e tamanho dos tecidos coletados. Embora o DNA do HPV seja detectado em CCEO, o seu papel etiológico permanece obscuro.2 Estudos anteriores demonstram que pacientes com tumores HPV positivos exibem uma maior taxa de sobrevida quando comparados a pacientes com tumores HPV negativos.19 Devido a esta observação, a identificação de HPV torna-se de suma importância clínica.
Uma abrangente meta-análise de 1982 a 1987 indica que o HPV é um fator de risco independente e de suma importância no CCEO, os resultados indicam que a detecção do HPV é duas a três vezes mais comum em lesões pré-malignas orais e 4,7 vezes mais comum em CCEO, quando comparado com a mucosa oral normal.12 A infecção por HPV parece ser independente da exposição ao álcool e ao tabaco, no entanto epidemiologicamente menor, pois a infecção por HPV apresenta menor prevalência que o consumo de álcool e tabaco.12
A maioria dos estudos não leva em consideração a idade dos indivíduos estudados, mas um importante estudo demonstra que a detecção de HPV foi maior em indivíduos jovens e do sexo masculino, sendo mais marcante em tumores bem-diferenciados.16 Todos os pacientes jovens não-fumantes e não-etilistas apresentavam tumores HPV positivos.16 Tais resultados suportam a teoria de que em adultos jovens o CCEO estaria ligado a vírus oncogênicos. Por sua vez, todos os pacientes extremamente jovens para o CCEO eram etilistas e 2 deles eram tabagistas moderados, e tal fato apresenta ainda a importância do efeito combinado da infecção por HPV com outros fatores de risco, como o consumo de tabaco e álcool.16 A maior prevalência de HPV em lesões bem diferenciadas pode ser determinada pelo ciclo de vida viral, que parece ser melhor suportado por lesões bem-diferenciadas.16
Algumas evidências sugerem que os HPVs de alto risco estão realmente envolvidos na carcinogênese oral: o HPV vem sendo diagnosticado em lesões de CCE oral, os ceratinócitos orais são transformados por HPV de alto risco via mecanismos envolvendo E6 e E7, e o HPV pode ser detectado tanto no tumor primário quanto em metástases, sendo que tais resultados foram obtidos em pesquisas de regiões geograficamente diversas.12
A definição do papel do EBV na carcinogênese oral é tão incerta quanto a do HPV, e enfrentamos a mesma dificuldade quanto à interpretação dos trabalhos devido aos mesmos fatores relacionados à amostra e à sensibilidade do método de análise. Entretanto, no caso do EBV, a situação é um pouco mais complicada. O EBV infecta tanto células epiteliais quanto linfócitos, e a maioria das análises não leva em conta se a presença é em células neoplásicas ou no infiltrado inflamatório adjacente ao tumor. Além disso, algumas análises por PCR (método mais sensível) e citologia esfoliativa não descartam a presença de saliva, que contém inúmeros linfócitos. Desta forma, a detecção de EBV em CCEO varia de 0 a 100%.21
Apesar disto, o EBV parece estar realmente implicado na carcinogênese oral, pois seus produtos, como a LMP1, por exemplo, são capazes de promover malignização in vitro.24 Além disso, a detecção de infecção latente por EBV em lesões pré-malignas e a expansão clonal provocada pelo EBV sugere que a infecção viral precede o desenvolvimento do tumor, podendo ser um evento inicial na carcinogênese oral.24 Outro importante achado é a associação entre a presença do EBV e elevado grau de atipia celular, pois os tumores bem-diferenciados apresentam um menor número de cópias do EBV, quando comparados com os tumores pouco-diferenciados.24
Por outro lado, o EBV pode ter sido levado às células neoplásicas pelo contato direto com linfócitos infectados, sendo a infecção permitida pelo baixo estado imunológico do paciente portador de neoplasia.22 Isto poderia ser muito comum, uma vez que 90% da população apresenta anticorpos para EBV.22
Uma vez que tanto o EBV quanto o HPV possuem produtos que atuam diretamente no ciclo celular e na ativação da apoptose, maior atenção deve ser dada a estes vírus, pois deve existir uma relação direta com o prognóstico. No caso do EBV, mais estudos para detecção de LMP-1 e BZLF-1 são necessários, pois tais proteínas podem interagir com a p53 impedindo a apoptose mediada por essa via, sendo esta via de fundamental importância para o tratamento por radioterapia e quimioterapia, que usualmente causa apoptose nas células malignas.23 Assim sendo, a presença destas proteínas diminuiria a eficácia do tratamento para o CCEO.23 No entanto, somente um estudo foi achado correlacionando o EBV ao prognóstico, mas não foram encontradas diferenças significativas entre os EBV+/; vale ressaltar que este trabalho detectou o DNA do EBV por PCR, não pesquisando a presença de LMP-1 ou BZLF-1.23 Já para o HPV, maior ênfase deve ser dada para a E6 e a P53, para que possamos saber se existe alguma influência no tratamento e prognóstico. Vale ainda ressaltar que não achamos na literatura nenhum trabalho que fizesse tal correlação. Podemos ainda considerar um efeito sinérgico entre o HPV e o EBV, que poderiam estar agindo conjuntamente na carcinogênese oral.
Por conseguinte a P53 continua sendo o gene mutado mais comumente encontrado.20 A p53 demonstra ter um papel fundamental na carcinogênese oral, pois apresenta freqüentes mutações e é extremamente sensível a todos os fatores de risco, como o HPV, o EBV, o tabaco, o álcool e a telomerase.20 Apresenta ainda um importante valor na determinação do prognóstico e da recorrência, sendo um útil marcador diagnóstico, podendo ser utilizado até mesmo para a avaliação de margens tumorais no tratamento loco-regional.20 No entanto, muitas pesquisas têm sido feitas para a avaliação de marcadores moleculares, mas a maioria conta com números pequenos de amostras, e uma extensa revisão indica que a P53 isoladamente não tem valor preditivo no prognóstico, mas as moléculas relacionadas ao P53 no controle da apoptose teriam uma papel fundamental na carcinogênese e deveriam ser avaliadas conjuntamente.30 Para uma melhor elucidação da interação da P53 com outros marcadores, um abrangente trabalho prospectivo seria necessário, na presença de alguns prováveis agentes etiológicos como os descritos acima, para que pudéssemos avaliar o papel dos mesmos no prognóstico do CCEO.
A telomerase vem sendo descrita como não detectável nas células e tecidos normais, mas é encontrada em células germinativas, imortalizadas e em células cancerígenas.7 A expressão de telomerase parece aumentar durante a progressão do CCEO, sendo menor em displasias e maior em lesões francamente invasivas, o que sugere uma relação íntima entre a atividade da telomerase e a carcinogênese oral.29 Os achados sugerem que a telomerase esteja envolvida em etapas precoces da carcinogênese oral.9,29 Assim sendo, a telomerase seria ativada geneticamente e representaria um evento crítico e inicial, que levaria então, à carcinogênese.9,29
Por outro lado, existe outra corrente que não acredita no modelo previamente estabelecido, afirmando que células normais possuiriam a capacidade de expressar telomerase em condições proliferativas.9 Desta forma, o uso da atividade da telomerase como um marcador geraria falsos positivos, dependendo do estado proliferativo celular em questão.9 Tais achados incluem ainda a observação de que células telomerase positivas apresentam telômeros menores que as com telomerase negativa, o que parece sugerir que existem outros meios de preservação do telômero que não a telomerase.9 Esta hipótese considera que todo epitélio, de contínua proliferação, seria telomerase positivo.29 Entretanto, existem outros fatores que devemos levar em consideração, algumas células teriam a habilidade de expressar telomerase, mas não podemos afastar a possibilidade de que tal células talvez representem stem cells do epitélio.29 Além disso, a presença de células telomerase positiva em tecidos normais ou áreas adjacentes a tumores pode representar a sinalização de eventos bioquímicos e moleculares, que ainda não tiveram expressão histológica.
Cabe ainda uma análise do método de detecção da telomerase, sendo o método mais utilizado o chamado TRAP, que significa protocolo de amplificação repetida telomérica. Embora seja o método mais sensível, não é o mais específico, pois amplifica toda a seqüência, e estudos demonstram que a seqüência mais específica seria a subunidade denominada hTERT.7 Algumas subunidades da telomerase são encontradas tanto em células normais quanto em células de CCEO, mas somente a hTERT demonstrou uma expressão distinta em CCEO.7 A diferenciação celular parece baixar a expressão da hTERT, mas não de outras subunidades, o que indica que a hTERT seria um importante marcador de diferenciação, ou de potencial de crescimento celular.7 Porquanto existe um aumento da atividade da telomerase no CCEO, este parece estar relacionado a, no mínimo, dois eventos na carcinogênese oral: o primeiro seria a perda de controle na estratificação e diferenciação do epitélio; e a segunda seria a super expressão individualizada de hTERT nas células.7
Apesar do aumento da compreensão sobre as alterações moleculares que levariam a célula à malignização, o papel destes fatores permanece obscuro. Uma imensa variedade de marcadores e possíveis agentes etiológicos é apresentada atualmente na literatura científica, o que demonstra a complexidade da carcinogênese oral, um evento multifatorial que requer a desestabilização de inúmeros sistemas de controle.30 Embora as pesquisas nesta área sejam exaustivas, incompletas, e muitas vezes frustrantes, estas são necessárias e devem ser estimuladas, pois a melhor compreensão da biologia celular e o exato conhecimento dos mecanismos moleculares críticos para a carcinogênese pode levar à uma terapia direcionada e, quem sabe, mais eficaz. Além disto, as variações individuais do curso clínico podem ser melhor compreendidas, o que auxiliaria na predição do prognóstico e na escolha terapêutica.
Comentários FinaisBaseado na informação atualmente disponível, pode ser afirmado que existem diferentes mecanismos responsáveis pela carcinogênese oral, de origem genética e/ou infecciosa que ainda pode variar de acordo com as diferenças geográficas, culturais, étnicas e sócio-econômicas.
Uma melhor definição sobre a etiologia e as alterações celulares relevantes à evolução desses tumores permitirá que estes sejam precocemente diagnosticados, adequadamente tratados e controlados, e futuramente prevenidos.
Referências Bibliográficas 1. Neville BW, Damm DD, Allen CM, Bouquot JE. Patologia Oral e Maxilofacial. 1a ed. Rio de Janeiro: Guanabara-Koogan; 1998. 705p.
2. Gillison ML, Koch WM, Capone RB et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. JNCI 2000; 92(9): 709-20.
3. Llewellyn CD, Johnson NW, Warnakulasuriya KAAS. Risk factors for squamous cell carcinoma of the oral cavity in young people - a comprehensive literature review. Oral Oncol 2001; 37(5): 401-18.
4. Llewellyn CD, Linklater K, Bell J et al. Squamous cell carcinoma of the oral cavity in patients aged 45 years and under: a descriptive analysis of 116 cases diagnosed in the south east of England from 1990 to 1997. Oral Oncol 2003; 39: 106-14.
5. Atula S, Grenman R, Laippala P, Syrjanen S. Cancer of tongue in patients younger than 40 years: a distinct entity? Arch Otolaryngol Head Neck Surg 1996; 122(2): 1313-9.
6. Veldman T, Horikawa I, Barrett JC, Schlegel R. Transcriptional activation of the telomerase hTERT gene by human papillomavirus type 16 E6 oncoprotein. J Virol 2001; 75(9):4467-72.
7. Fujimoto R, Kamata N, Yokoyama K et al. Expression of telomerase components in oral keratinocytes and squamous cell carcinomas. Oral Oncol 2001; 37: 132-40.
8. Scully C. Oral squamous cell carcinoma; from an hypothesis about a virus, to concern about possible sexual transmission. Oral Oncol 2002; 38: 227-34.
9. Belair CD, Yeager TR, Lopea PM, Reznikoff CA. Telomerase activity: a biomarker of cell proliferation, not malignant transformation. Proc Natl Acad Sci USA 1997; 94:13677-82.
10. Lydiatt D. Cancer of the oral cavity and medical malpractice. Laryngoscope 2002; 112(5): 816-9.
11. Oliver RJ, Dearing J, Hindle I. Oral cancer in young adults: report of three cases and review of the literature. Br Dent J 2000; 188(7): 362-6.
12. Miller CS, Johnstone, BM. Human papillomavirus as a risk factor for oral squamous cell carcinoma: a meta-analysis, 1982-1997.Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2001; 91(6): 622-35.
13. Lype EM, Pandey M, Mathew A et al. Oral cancer among patients under the age of 35 years. J Postgrad Med 2001; 47(3):171-6.
14. Wattre P. Apoptose et virus (hormis les retrovirus). Revue Française des Laboratoires 1999; 311: 43-9.
15. Bouda M, Gorgoulis VG, Kastrinakis NG et al. "High risk" HPV types are frequently detected in potentially malignant and malignant oral lesions, but not in normal oral mucosa. Mod Pathol 2000; 13: 644-53.
16. Cruz IBF, Snijders PJF, Steenbergen RDM et al. Age-dependence of human papillomavirus DNA presence in oral squamous cell carcinomas. Oral Oncol 1996; 32B (1): 55-62.
17. Mork J, Lie AK, Glattre E et al. Human papillomavirus infection as a risk factor for squamous cell carcinoma of the head and neck. N Engl J Med 2001; 344(15): 1125-31.
18. Premoli-De-Percoco G, Ramirez JL, Galindo I. Correlation between HPV types associated with oral squamous cell carcinoma and cervicovaginal cytology: an in situ hybridization study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1998; 86(1): 77-81.
19. Capone RB, Pai SI, Koch WM et al. Detection and quantitation of human papillomavirus (HPV) DNA in the sera of patients with HPV - associated head and neck squamous cell carcinoma. Clin Cancer Res 2000; 6(11): 4171-5.
20. Gasco M, Crook T. The p53 network in head and neck cancer. Oral Oncol 2003; 39: 222-31.
21. Sand LP, Jalouli J, Larson P-A, Hirsch J-M. Prevalence of Eptein-Barr virus in oral squamous cell carcinoma, oral lichen planus, and normal oral mucosa. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2002; 93(5): 586-92.
22. Cruz I, Van Den Brule AJ, Brink AA et al. No direct role for Epstein-Barr virus in oral carcinogenesis: a study at the DNA, RNA and protein levels. Int J Cancer 2000; 86: 356-61.
23. Maeda T, Hiranuma H, Matsumura S, Furukawa S, Fuchihata H. Epstein-Barr virus infection and response to radiotherapy in squamous cell carcinoma of the oral cavity. Cancer Letters 1998; 125:25-30.
24. Gonzalez-Moles MA, Gutierrez J, Rodriguez MJ, Ruiz-Avila I, Rodriguez-Archilla A. Epstein-Barr virus latent membrane protein - 1 (LMP-1) expression in oral squamous cell carcinoma. Laryngoscope 2002; 112(3): 482-7.
25. Fields BN, Knipe DM, Howley PM. Fields Virology. Philadelphia: Lippincott-Raven Publishers; 1996.
26. Hsieh LL, Wang P-F, Chen IW et al. Characteristics of mutations in the p53 gene in oral squamous cell carcinoma associated with betel quid chewing and cigarrete smoking in Taiwanese. Carcinogenesis 2001; 22(9): 1497-503.
27. Gonzalez-Moles MA, Galindo P, Gutierrez J et al. Expression of the p53 protein in oral squamous cell carcinomas associated with Epstein-Barr virus. Microbios 2000; 102(403):147-54.
28. Optiz OG, Suliman Y, Hahn WC et al. Cyclin D1 overexpression and p53 inactivation immortalize primary oral keratinocytes by a telomerase-independent mechanism. J Clin Invest. 2001; 108(5):725-32.
29. Liao J, Mitsuyasu T, Yamane K, Ohishi M. Telomerase activity in oral and maxillofacial tumors. Oral Oncol 2000; 36: 347-52.
30. Schliephake H. Prognostic relevance of molecular markers of oral cancer - a review. Int J Oral Maxillofac Surg 2003; 32: 233-45.
1 Mestranda em Patologia Bucodental da UFF, Especialista em Estomatologia pela UFRJ.
2 Professora Adjunta do Departamento de Patologia e Diagnóstico Oral da FO/UFRJ. Doutora em Patologia Bucal pela USP.
3 Professora Adjunta do Departamento de Patologia do Hospital Universitário Antônio Pedro da Universidade
Federal Fluminense. Doutora em Patologia Bucal pela FOUSP-Bauru.
Departamento de Patologia da Universidade Federal Fluminense - Niterói / RJ.
Curso de Pós Graduação - Mestrado em Patologia Bucodental.
Endereço para Correspondência: R. Soldado Francisco de Souza, 110 Rio de Janeiro RJ 22770-155.
Tel (0xx21) 3392-0418 - E-mail: beatriz_venturi@ig.com.br
Artigo recebido em 07 de dezembro de 2003. Artigo aceito em 18 de fevereiro de 2004.
