IntroduçãoO estudo da surdez de origem genética enumera possibilidades diagnósticas cada vez mais amplas. Muitos pacientes com perda auditiva sensório-neural de causa indefinida podem apresentar alterações cromossômicas determinantes de sua patologia.
A realização de exames simples e acessíveis para investigar as mutações mais comuns associadas a perdas auditivas tornou possível o esclarecimento etiológico de alguns casos.
Calcula-se que mais de 100 genes estejam potencialmente envolvidos na deficiência auditiva não-sindrômica. Após a localização do locus DFNA1 no cromossomo 5q31 em 1992, cerca de 70 loci diferentes já foram mapeados. Desde o relato da primeira mutação em gene nuclear associada à surdez autossômica não-sindrômica em 1997, outros 15 genes foram clonados.1,2
Estima-se que 70% de todas as causas genéticas de surdez sejam não-sindrômicas e entre estas, 80% se apresentam com padrão de herança autossômico recessivo. O gene GJB2, que codifica a proteína conexina 26 (Cx26), está envolvido tanto nas formas dominantes quanto recessivas de surdez não-sindrômica. Mutações neste gene são responsáveis por 80% dos casos com padrão de herança recessivo e uma mutação específica (35delG) é a maior envolvida nos casos de surdez com etiologia genética. Trata-se da perda de uma base guanina da seqüência de DNA deste gene na posição 35. Esta mutação corresponde de 75 a 80% daquelas encontradas neste gene.3,4
A mutação A1555G no gene mitocondrial 12SrRNA foi a primeira associada com surdez não-sindrômica e determina um padrão de herança materno5. Esta mutação tem sido associada à surdez vinculada a ototoxicidade dos aminoglicosídeos e nos Estados Unidos ela está presente em 15% de todos os pacientes com surdez induzida por estes antibióticos. Outra mutação mitocondrial que determina surdez é a A7445G no gene tRNASer (UCN) que, em alguns casos, está associada a ceratodermia palmoplantar6,7.
ObjetivosInvestigar a presença das mutações 35delG/GJB2, A1555G/12SrRNA e A7445G/tRNASer (UCN) em pacientes cujo diagnóstico etiológico para a perda auditiva não tenha sido determinado ou que apresentem dados positivos no heredograma. Avaliar a influência destas informações na definição etiológica da deficiência auditiva.
Material e MétodosForam incluídos neste trabalho os pacientes atendidos nos Ambulatórios de Otoneurologia e Distúrbios da Comunicação da Disciplina de Otorrinolaringologia-Cabeça e Pescoço da Universidade Estadual de Campinas - UNICAMP, de julho a dezembro de 2000, que satisfizessem pelo menos um dos seguintes critérios:
1. Perda auditiva sem diagnóstico etiológico conclusivo.
2. Perda auditiva desproporcional à suposta causa identificada.
3. Perda auditiva precoce.
4. História familial positiva ou existência de consangüinidade.
Os pacientes incluídos em algum dos critérios definidos foram submetidos à avaliação genética, independente da realização de outros exames indicados (perfil metabólico, perfil auto-imune, provas sorológicas, exames por imagem de alta resolução).
As amostras de sangue foram colhidas após esclarecimento dos objetivos do teste e assinatura do termo de consentimento. Foram enviadas ao Laboratório do Centro de Biologia Molecular e Engenharia Genética da Universidade Estadual de Campinas (CBMEG), procedendo-se à pesquisa da mutação 35delG da Cx26 e das mutações mitocondriais A1555G e A7455G nos genes 12SrRNA e tRNASer (UCN) respectivamente. Em seguida, o seqüenciamento estendeu-se à procura de outras mutações no GJB2. Identificada qualquer alteração, o acompanhamento clínico passava a ser realizado também pelo Departamento de Genética da Faculdade de Ciências Médicas - UNICAMP.
Todas as amostras de DNA foram triadas para a presença da mutação 35delG utilizando a técnica reação em cadeia da polimerase alelo-específica (AS-PCR), porém com modificação patenteada pelo CBMEG - UNICAMP. Este método discrimina facilmente o alelo normal do mutante e através de duas reações é possível distinguir homozigotos normais, homozigotos 35delG e heterozigotos portadores da mutação8.
Para investigar as mutações mitocondriais foram amplificadas pela técnica PCR duas regiões do DNA mitocondrial de cada paciente, uma contendo o nucleotídeo 1555 e outra contendo o nucleotídio 7445, de acordo com a publicação de Cambridge de seqüência mitocondrial9.
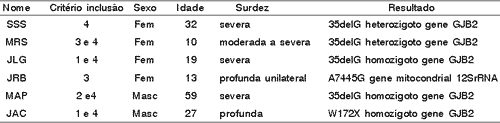
ResultadosNos 75 pacientes que preencheram os critérios propostos foram detectadas seis mutações, sendo quatro indivíduos portadores da mutação 35delG (Quadro 1). Destes, dois são homozigotos e dois são heterozigotos para a mutação. Das outras duas mutações encontradas, uma situa-se também no GJB2 (homozigoto W172X/W172X) e é uma nova mutação nunca descrita. A outra (A7445G) foi identificada no gene mitocondrial 12SrRNASer (UCN). Em 19 pacientes está concluído o seqüenciamento total do GJB2 e destes, 14 são normais. Em 56 pacientes não foi evidenciada a presença da 35delG mas outras mutações, relacionadas ou não a Cx26, ainda podem ser encontradas. 73 pacientes não tinham as mutações mitocondriais investigadas.
Caso 1
MRS, feminino, 10 anos, hipoacusia neurossensorial bilateral moderada/severa progressiva há 3 anos, acompanhada durante 8 meses com diagnóstico de otoesclerose coclear. Sua avaliação inicial baseava-se no antecedente familial de perda auditiva materna, também considerada otoesclerose. Resultado do exame genético: heterozigose para 35delG.
Caso 2
MAP, masculino, 59 anos, hipoacusia neurossensorial bilateral severa desde criança, com antecedentes de otorréia na infância. 3 irmãos com perda auditiva. Resultado do exame genético: homozigose para 35delG.
Caso 3
SSS, feminino, 32 anos, hipoacusia neurossensorial bilateral severa com pais consangüíneos, irmão e filha com perda auditiva. Acompanhada por quase 4 anos com diagnóstico de otoesclerose coclear. Resultado do exame genético: heterozigose para 35delG.
Caso 4
JLG, feminino, 19 anos, hipoacusia neurossensorial bilateral severa desde infância, com histórico familial de 3 irmãos com surdo-mudez. Resultado do exame genético: homozigoze para 35delG.
Caso 5
JAC, masculino, 27 anos, hipoacusia neurossensorial bilateral severa a profunda desde a infância. 9 irmãos, 3 com surdez sendo 2 destes também desde a infância. Resultado do exame genético: homozigose para W172X.
Caso 6
JRB, feminino, 15 anos, hipoacusia profunda esquerda, leve a moderada direita. Rubéola materna e positividade sorológica ao nascer segundo informações maternas. Antecedentes de otites de repetição e uso de drenos de ventilação bilateral. Resultado do exame genético: A7445G gene mitocondrial 12SrRNA.
Quadro 1. Mutações encontradas na investigação genética realizada.
As conexinas são proteínas constituintes das junções intercelulares, local primordial de comunicação entre as células para trocas de eletrólitos, mensageiros secundários e metabólitos. A Cx26 oligomeriza com outras cinco unidades idênticas (homotípicas) ou não idênticas (heterotípicas) formando hexômeros chamados conexons. Estes, embebidos na membrana plasmática, formam pontes dissulfídicas com conexons das células vizinhas, facilitando transferências diretas de pequenas moléculas de citoplasma a citoplasma10. No ouvido interno, a Cx26 influencia a função da estria vascular, membrana basilar, limbus e proeminência espiral da cóclea humana. A perda de função desta conexina neste complexo contribui para alterar a permeabilidade das células de suporte e fibroblastos do órgão de Corti, permitindo que o potássio presente em grande concentração no ducto coclear se difunda no órgão, modifique o transporte e permeabilidade deste íon nas sinapses das células ciliadas e acarrete a perda auditiva11.
Em avaliação microscópica do osso temporal de um paciente com surdez que apresentava mutação 35delG heterozigótica, Jun et al. evidenciaram ausência de degeneração neural, células do gânglio espiral preservadas, degeneração quase total das células ciliadas do órgão de Corti, membrana tectorial desconectada, agenesia da estria vascular e um grande cisto na escala média na região da estria vascular12.
Não foi determinada ainda uma correlação entre a mutação específica 35delG e a severidade ou progressão da perda auditiva. Cohn et al. não encontraram nenhum fenótipo auditivo consistente para alterações no locus DFNB13. Rabionet et al. sugerem que estas variações indicariam a participação de fatores ambientais13. Park et al. propõem que manifestações diferenciadas das mutações da Cx26 devam ser atribuídas ao fator racial14. Desta forma, apesar da identificação destas anomalias fornecerem informações importantes para aqueles que já apresentam surdez, é preciso muita cautela em qualquer afirmação referente aos pacientes que possuem a variação genética sem doença demonstrável. No entanto, existindo o risco de 25% de recorrência do genótipo, fica evidente a importância do aconselhamento.
Apesar de o genótipo não predizer a audição, homozigotos para 35delG têm incidência de surdez pré-lingual significativa: 26 a 30% terão surdez severa e outros 30 a 57% profunda. Perda auditiva severa a profunda afeta aproximadamente 1 para 1000 neonatos. Em 50% destas crianças, a perda é presumivelmente genética, herdada de forma autossômica recessiva. Por esta razão, sugere-se que o teste genético, pelo seu baixo custo, deva ser incluído na bateria de testes na investigação de perdas auditivas neurossensoriais4,11,12.
Em nosso estudo, quatro pacientes apresentaram teste genético positivo para a 35delG, sendo que dois são heterozigotos, dois homozigotos e todos apresentavam perda auditiva severa.
A presença de outras condições mórbidas pode alterar a situação clínica do paciente modificando as manifestações da doença genética em si. Dos nossos seis pacientes mutantes, um (SSS) apresentava Diabetes Melitus tipo II compensado e os antecedentes de outro (JRB) mascaravam a origem genética da alteração auditiva.
Alguns autores acreditam que outras mutações envolvendo a Cx26 podem estar relacionadas à fisiopatologia de algumas doenças degenerativas do ouvido e explicariam perdas auditivas progressivas como a presbiacusia15.
Muitos genes associados à surdez hereditária, ao contrário do que se supunha, não se expressam nas células ciliadas mas sim nas células de suporte distribuídas ao longo do ducto coclear. A descoberta de mutações e seus efeitos na audição veio mostrar que o papel destas células na função coclear é indispensável1.
Embora existam controvérsias, o envolvimento do GJB2 em surdez autossômica dominante também foi proposto em pequeno percentual destes casos (foram descritas 5 destas mutações)16-18.
Poucos artigos relacionando mutações associadas à perda auditiva e função vestibular foram publicados. Os pacientes apresentados neste trabalho não apresentaram anormalidades vestibulares clínicas. Kunst et al. descrevem alguns achados vestibulares instrumentais de caráter inespecífico e sem padrão identificável2,12,13.
A incidência das mutações no gene GJB2 na literatura mundial é superior aos dados obtidos em nossa casuística, o que se atribui ao critério rigoroso de seleção dos pacientes e ao fato de ainda não haver sido finalizado o seqüenciamento completo do gene de todos os pacientes nesta investigação.
A mutação W172X em homozigose, como observada no paciente JAC, nunca foi relatada anteriormente na literatura. Foi confirmada por análise de restrição e provoca a troca do aminoácido triptofano por codon de terminação interferindo claramente na função da proteína. Esta mutação foi descrita somente neste paciente brasileiro com provável consangüinidade na família.
Mitocôndrias são organelas intracelulares responsáveis pela produção de maior parte da energia produzida nas células. Elas possuem um pequeno conteúdo genômico com padrão materno de herança que, quando mutante, pode originar um largo espectro de doenças19. Mutações no DNA mitocondrial são associadas tanto à surdez não-sindrômica quanto sindrômicas. Fatores ambientais como os antibióticos aminoglicosídeos, assim como prováveis genes nucleares ainda não-identificados, interagiriam com estas mutações determinando a expressão dos fenótipos auditivos19,20.
Foram descritas cinco mutações determinando perda auditiva não-sindrômica. Em duas destas mutações (7445G, 7472C), além da deficiência auditiva, outros sintomas podem estar presentes. Desconhece-se porque estas cinco mutações preferencialmente afetam o ouvido interno, apesar do papel fundamental que as mitocôndrias exercem em quase todas as células do corpo humano19.
A manifestação fenotípica das doenças decorrentes de alterações no DNA mitocondrial é comumente associada a início tardio, seletividade de órgão atingido e uma evolução progressiva em crises21.
ConclusãoAtualmente a triagem das mutações genéticas associadas à perda auditiva é de fácil realização e custo acessível.
A mutação 35delG da Cx 26 está potencialmente vinculada a alguns casos de perda auditiva não esclarecida. A pesquisa desta mutação deve ser incluída na bateria de exames de investigação etiológica da surdez indeterminada, uma vez que auxilia na elucidação da causa e possibilita, em caso de positividade, o aconselhamento genético.
Em pacientes nos quais exista grande suspeição de doença hereditária deve-se prosseguir o estudo em outros genes.
Referências Bibliográficas 1. Steel KP, Bussoli TJ. Deafness genes, expressions of surprise. TIG 1999; 15(6):207-11.
2. Kunst H, Marres H, Huygen P, Van Duijnoven G, Krebsova A, Van Der Velde S, Reis A, Cremers F, Cremers C. Non-syndromic autosomal dominant progressive non-specific mid-frequency sensorineural hearing impairment with childhood to late adolescence onset (DFNA21). Clin Otolaryngol 2000;25:45-54.
3. Cohn ES, Kelley PM, Fowler TW et al. Clinical studies of families with hearing loss attributable to mutations in the connexin gene (GJB2/DFNB1). Pediatrics 1999; 103(3): 546-50.
4. Cohn ES, Kelley PM. Clinical phenotype and mutations in connexin 26 (DFNB1/GJB2), the most commun cause of childhood hearing loss. Am J Med Genet 1999; 89:130-6.
5. Jaber L, Shohat M, Bu X, Fischel-Ghodsian N, Yang HY, Wang SJ, Rotter JI. Sensorial deafness inherited as a tissue specific mitochondrial disorder. J Med Genet. 1992; 29:86-90.
6. Fischel-Ghodsian N, Prezant TR, Fournier P, Stewart IA, Maw M. Mitochondrial tRNA mutation associated with non-syndromic deafness. Am J Otolaryngol 1995; 16: 403-8.
7. Sevior KB, Hatamochi A, Stewart IA et al. Mitochondrial A7445G mutation in two pedigrees with palmoplantar keratoderma and deafness. Am J Med Genet 1998; 75:179-85.
8. Sartorato CA, Gottardi E, Oliveira CA et al. Determination of carrier frequency of the 35delG mutation in Brazilian neonates. Clinical Genetics 2000; 58(4): 339.
9. Anderson S, Bankier AT, Barrel BG et al. Sequence and organization of the mitochondrial genome. Nature 1981; 290:457-65.
10. Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem 1996; 238(1):1-27.
11. Lefebvre PP, Van De Water TR. Connexins, hearing and deafness: clinical aspects of mutations in the connexin 26 gene. Brain Research Reviews. 2000; 32:159-62.
12. Jun AI, Mcguirt WT, Hinojosa R, Green GE, Fischel-Ghodsian N, Smith RJH. Temporal bone histopathology in Connexin 26 related hearing loss. Laryngoscope 2000; 110:269-75.
13. Rabionet R, Gasparini P, Estivill X. Molecular genetics of hearing impairment due to mutations in gap junction genes encoding beta connexins. Human Mutations 2000; 16:190-202.
14. Park HJ, Hanh SH, Chun YM, Park K, Kim H. Connexin 26 mutations associated with nonsyndromic hearing loss. Laryngoscope 2000; 110:1535-8.
15. Morell RJ, Kim HJ, Hood LJ et al. Mutations in the connexin 26 gene (GJB2) among Ashkenazi jews with nonsyndromic recessive deafness. N Engl J Med 1998; 339:1500-5.
16. Denoyelle F, Lina-Granade G, Plauchu H et al. Connexin 26 gene linked to a dominant deafness (letter). Nature 1998; 393:319-20.
17. Scott DA, Kraft ML, Stone EM, Sheffield VC, Smith RJ. Connexin mutations and hearing loss (letter). Nature 1998; 391:32.
18. The Connexin-deafness homepage. http://www.iro.es/cx26.htm
19. Van Camp G, Smith RJ. Maternally inherited hearing impairment. Clin Genet 2000 Jun; 57(6):409-14.
20. Jacobs HT. Mitochondrial deafness. Ann Med 1997 Dec; 29(6):483-91.
21. Guan MX, Fischel-Ghodsian N, Attardi G. Biochemical evidence for nuclear gene involvement in phenotype of non-syndromic deafness associated with mitochondrial 12S rRNA mutation. Hum Mol Genet 1996 Jul; 5(7):963-71.
1 Médico Assistente dos Setores de Otoneurologia e Base do Crânio da Disciplina de Otorrinolaringologia e Cirurgia de Cabeça e Pescoço- UNICAMP.
2 Médica Assistente do Setor de Otoneurologia da Disciplina de Otorrinolaringologia e Cirurgia de Cabeça e Pescoço-UNICAMP.
3 Doutor em Genética Humana - UNICAMP-CBMEG.
4 Ex-residente da Disciplina de Otorrinolaringologia e Cirurgia de Cabeça e Pescoço- UNICAMP.
5 Doutor em Genética Humana- UNICAMP- FCM Cidade Universitária ZeferinoVaz
Trabalho apresentado no II Congresso Triológico de Otorrinolaringologia realizado em Goiânia em 20001 e foi classificado recebendo Menção Honrosa.
Disciplina de Otorrinolaringologia e Cirurgia de Cabeça e Pescoço - UNICAMP
Endereço para Correspondência: Cidade Universitéria Zeferino Vaz CP 6111, Barão Geraldo, Campinas, SP 13083-970
Tel (0xx19) 3788-7523 - E-mail: lnp@uol.com.br
Artigo recebido em 01 de agosto de 2003. Artigo aceito em 04 de setembro de 2003.
