IntroduçãoSamelsohn(1) em 1874 descreveu pela primeira vez casos de nefrite hereditária mas nada observou sobre surdez Dickinson(2) verificou albuminuria em 11 casos de 17 membros de uma família. Foi Cecil Alport(3) em 1927 o primeiro autor a descrever a história de uma família com surdez e nefropatia. Desde então esta síndrome foi denominada de síndrome de Alport. Rinkoff(4) descreveu mais tarde a mesma síndrome e observou que havia surdez neurosensorial e que a progressão da nefropatia era pararela com a lesão auditiva. Sohar(5) notou em 1954 que junto haviam lesões oculares, principalmente catarata, discromatopsia e miopia. A finalidade deste trabalho é apresentar o caso de uma família com síndrome de Alport com estudo anátomo patológico de um dos membros que faleceu.
Estudo de uma Família
Trataremos de 7 irmãos, seis homens e 1 mulher, com idades que variam de 9 a 21 anos, filhos de mãe e pai sadios, mas com antecedentes de doença renal e suspeita de surdez (quadro I). O quadro mostra o que foi possível levantar genéticamente sobre o grupo familiar dos antecedentes da família em estudo. Todos nasceram de parto normal e com desenvolvimento razoável, até que se começou a notar em alguns membros problemas os mais diversos. Submetidos a exame com o nefrologista constatou-se que 4 dos filhos masculinos eram portadores de proteinúria intensa, hematúria e cilindrúria (quadro II). O audiograma revelou disacusia neurosensorial em três elementos coincidindo a alteração auditiva com os que eliminavam mais proteínas pela urina (Fig. nº 1, 2, e 3), os demais apresentavam audição normal. Foram controlados periodicamente pelo nefrologista e pelo médico otorrinolaringologísta por vários anos e até hoje são submetidos periodicamente a exames. No mês de setembro de 1967 um dos irmãos sofreu um agravamento do estado geral, tendo baixado em 31.7.68 no Hospital Ernesto Dornelles para avaliação clínica, para receber transfusão de sangue e para ser submetido a punção-biópsia renal. O paciente vem sendo controlado semanalmente há 2 meses, clínica e laboratorialmente, tendo apresentado uma tensão arterial que se mantém em torno de 170/100. A proteinúria tem se mantido em torno de 5 g em 24 horas e a depuração da creatinina e andágena em torno de 11. O paciente apresenta, desde o início do controle clínico-laboratorial, as mucosas persistentemente descoradas. Apesar disso, sente-se bem, não aceitando entretanto a sua doença. Aproximadamente há 1 mês, vem tomando Meticorten, 200 mg em dias alternados, não tendo apresentado melhoras. A tensão arterial é 170/105. 0 psiquismo é lúcido. A temperatura axilar 36,5 e o pulso 100 batimentos por minuto. Atitude ativa indiferente. Estado de nutrição bom. Pele e mucosa descoradas. Ausculta pulmonar sem particularidades. Ausculta cardíaca com ritmo normal, 98 batimentos por minuto, sobre sistólico + audível em todos os focos. Fígado e baço impalpáveis. Nitrogênio uréico 32,5 mg%, uréia 69,5 mg%, creatinina 8,0 mg%, potássio 5 mili equivalentes por litro, C02-16 mili equivalentes por litro. Eritrócitos 3,5 milhões por mm3, hemoglobina 10,15 g% (70%), hematócrito 31,0%, valor globular 1. Em 15.8.68: Nitrogênio uréico 40 mg%, uréia 85,6%, creatinina 7,1 mg%. Em 12.8: nitrogênio uréico 46,4 mg%, uréia 99,2 mg%. Em 1.8.: eritrócitos 2,2 milhões por, mm3, hemoglobina 6,09%, hematócrito 19%, leucócitos 7700 mm3, com 6 bastonetes, 84 segmentados, 4 monócitos e 16 linfócitos. Plaquetas 200 000/mm3. 2ª baixa: Em 13.9.1968, por nefrite crônica familiar acompanhada de surdez (Síndrome de Alport). Há um ano, entrou em insuficiência renal, evoluindo rapidamente para a fase urêmica avançada. Há uma semana, a uréia elevou-se a 280 mg%. Vem vomitando com insistência e apresentando sangramento difuso da faringe e gengivas, anorexia, anemia profunda e edema palpebral. Nitrogênio uréico 96 mg% e uréia 204,8 mg%, eritrócitos 3,57 milhões por mm3. Hemoglobina 10,15 g%, hematócrito 31%, leucócitos 9000/mm3 com 10 bastonetes, 75 segmentados, 7 monócitos e 8 linfócitos. Em 12.10: o nitrogênio uréico elevou-se para 106,4 mg% e a uréia para 227, 2 mg%. A cultura de liquido peritoneal revelou Klebsiella, sensível a kanamicina e novabiocina. O estado do paciente continuou agravando-se, conservou-se sonolento, com vômitos freqüentes e diarréia. Recebeu várias transfusões de sangue total e, foi submetido a diálises repetidas. Entrou em coma em 13.10, piorando cada vez mais o seu estado geral. O óbito ocorreu às 3,50 hs. do dia 20.10. Protocolo: A necrópsia é realizada no cadáver de um jovem branco, em mau estado de nutrição, bem constituído, com 17 anos de idade. A pele não apresenta ferimentos ou cicatrizes. Os gânglios linfáticos superficiais são impalpáveis. Mamas sem alterações. Tecido adiposo subcutâneo: Mede 4 mm ao nível da cicatriz umbilical. Músculos: sem alterações. Cavidade peritonial: Contém cerca de 1 litro de líquido turvo e pardo-amarelado. Retro-peritôneo: Sem alterações. Margem do fígado: Esta situada a 7,0 cm abaixo da ponta do apêndice xifóide e a 1,0 cm acima da margem costal direita, na linha hemi-clavicular. O diafragma eleva-se ao nível do 4.° espaço intercostal em ambos os lados. Faringe: Sem alterações. Esôfago: Sem alterações. Estômago: Contém cerca de 100 ml de substância líquida, turva e de cor verde. A mucosa não apresenta alterações. Intestinos: Acham-se difusamente aderentes, sendo a serosa recoberta por membrana de aspecto fibrino-purulento. A abertura, contém fezes líquidas. A mucosa não apresenta alterações. Apêndice cecal: sem alterações. Mesentério: Gânglios linfáticos e vasos sanguíneos sem alterações. Laringe: Sem alterações. Tireóide: Pesa 17,0 g e a superfície de corte não apresenta alterações. Paratireóide: Foram dissecadas as duas paratireóides superiores que não apresentam alterações macroscópicas. Mediastino: Sem alterações. Timo não foi dissecado. Cavidades pleurais: Ambas contêm mínima quantidade de líquido de cor amarelocitrino. Traquéia-Bronquicos: Sem alterações. Gânglios linfáticos bronquiais: Não apresentam alterações. Pulmões: Pesam junto 500,0 g e estão em colapso parcial. A superfície de corte é pouco arejada, seca e de cor rósea. Artéria pulmonar: Não apresenta alterações. Pericárdio: Contém cerca de 100 ml de líquido amarelo-citrino. Coração: Pesa 450,0 g. O epicárdio não apresenta alterações. O ventrículo direito mede 3 mm de espessura e a parede do ventrículo esquerdo mede 22 mm de espessura. As colunas carnosas do ventrículo esquerdo estão espessadas. As cavidades ventriculares e a aurícula esquerda são distendidas. O endocárdio não apresenta alterações. O foramen oval está fechado. A válvula mitral mede 9,0 cm de circunferência, a válvula aórtica 6,5 cm, a válvula tricúspide 11,0 cm e a válvula pulmonar 8,0 cm de circunferência. As artérias coronárias não apresentam alterações. Aórta: Sem alterações. Veia cavatributárias: Sem alterações. Fígado: Pesa 1 800 g e a cápsula está parcialmente recoberta por membrana de cor amarela. A superfície de corte é marcadamente congesta e de cor pardaavermelhada. A artéria e veia hepática não apresentam alterações. Veia porta: Sem alterações. Vesícula biliar: Contém cerca de 10 ml de bile verde-amarelada. Ductos filiares: Sem alterações. Pâncreas: Pesa cerca de 40,0 g, a superfície de corte não apresenta alterações. Baço: Pesa 250,0 g e a cápsula está parcialmente recoberta por membrana amarela. A superfície de corte é vermelho-vinhosa e apresenta folículos proeminentes. A artéria e veias esplênicas não apresentam alterações. Supra-renais: Pesam 6,5 g cada uma. Ao corte a superfície não apresenta anormalidades. Rins: O direito pesa 75 g e o esquerdo pesa 70 g e medem 8,5 x 4,5 x 3,0 cm. As cápsulas destacamse com relativa facilidade e a superfície capsular é uniformemente granular e congesta, medindo os grânulos de 1 a 2 mm de diâmetro. Há persistência de lobulação fetal. Ao corte a cortical mede 2 a 3 mm de espessura e a medular 0,8 a 1,0 cm. As cavidades pielocalicinais não apresentam alterações. Ureteres; Sem alterações. Bexiga: Contém cerca de 100 ml de urina sem alterações macroscópicas. Uretra: Sem alterações. Pênis: Sem alterações. Próstata: Sem alterações. Vesículas seminais: Sem alterações. Testículo: Sem alterações. Cérebro: Pesa 1300 g. Foram retirados os ossos temporais de ambos os lados.
QUADRO I Grupo familiar dos antecedentes dos 7 irmãos estudados. Está assinalado o membro que foi estudado anatomopatológicamente após o falecimento.
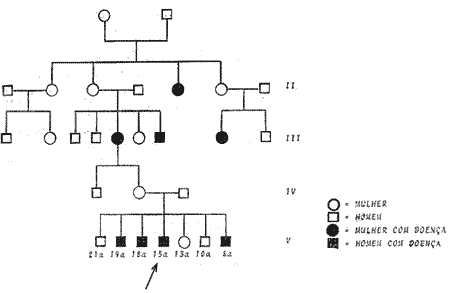
QUADRO II Dados das alterações urinárias dos 7 irmãos em estudo, onde vemos que 4 são portadores de hematúria, proteinúria e cilindrúria.
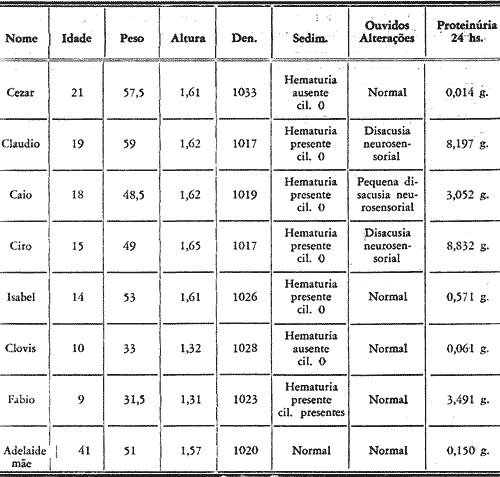
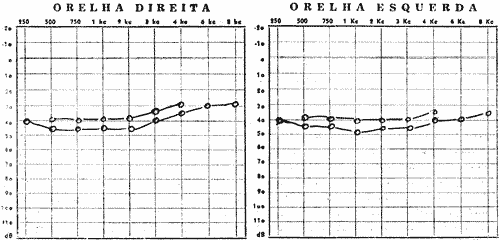
FIG. Nº 1 - (A e B) Audiograma do irmão nº 2, na cronologia (Cláudio) o qual apresenta disacusia neurosensorial bilateral com sisi positivo nas freqüências 2k, 3k e 4k.
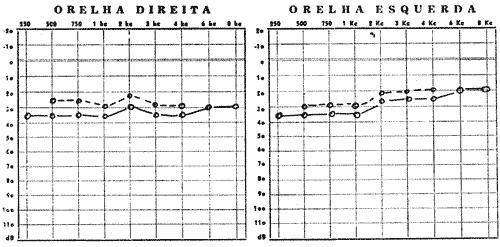
FIG. Nº 2 - (A e B) Audiograma do irmão nº 3, na cronologia (Caio) com disacusia neurosensorial bilateral e sisi com positividade de 92% em 2k, 3k e 4k. Indicando a existência de lesão coclear.
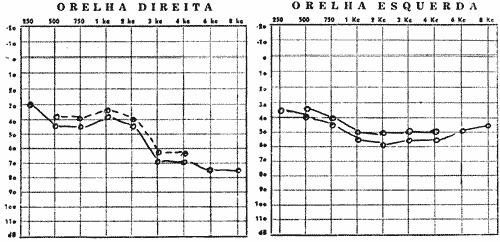
FIG. Nº 3 - (A e B) Audiograma do irmão nº 4 na cronologia (Ciro) com disacusia neurosensorial e sisi positivo 62% nas posições 2k, 3k e 4k.
Exame Microscópico
Coração: Há hipertrofia de fibras miocárdicas. Pulmões: Poucos alvéolos contêm fibrina e macrófagos com hemossiderina. Há colapso focal. Baço: Os sinusóides e espaços inter-sinusoidais são marcadamente congestos. Há, na polpa, linfócitos, plásmócitos, neutrófilos e numerosos macrófagos contendo hemossiderina. A cápsula esta recoberta por exsudato neutrocitário. Fígado: Os sinusóides são congestos. Os espaços portais apresentam discretos infiltrados linfóides. A cápsula está recoberta por exsudato neutrocitário. Pâncreas: Ausência de alterações histológicas. Supra-renais: Há congestão da camada reticular. Rins: A maior parte dos glomérulos apresenta alterações marcadas, como sejam espessamento do mesângio, sinéquias, proliferação endotelial, crescentes epiteliais e fibrosos, fibrose capsular e esclerose de tufo. Alguns glomérulos esparsos estão bem conservados. Há acentuada atrofia tubular, com áreas de túbulos hipertrofiados e dilatados, alguns contendo cilindros proteináceos e neutrófilos. As células tubulares apresentam, por vezes, citoplasma vacuolizado por prováveis lipídios. Raras trabéculas contêm cristais de cálcio. O estroma apresenta fibrose focal e infiltrados linfo-plasmocítários. Neste, identificam-se agrupamentos esparsos de células de citoplasma espumoso ("Foam Cells"). As artérias musculares revelam espessamento acentuado da íntima com edema. Esôfago: Ausência de alterações histológicas. Estômago: A serosa está recoberta por exsudato neutrocitário. Intestinos: A serosa está recoberta por exsudato neutrocitário. Testículo: O epitélio germinativo está conservado, não havendo espermatogênese. Tireóide: Ausência de alterações histológicas. Medula óssea: Há muitos neutrófilos e hemácias nucleadas.
ComentáriosEste paciente apresentou os achados patológicos renais característicos de nefrite hereditária, conforme a descrição de Krickstein et col.(6) Células espumosas ("Foam Cells'") foram demonstradas nos túbulos e estroma. Este tem sido interpretado como achado peculiar, quando associado, como no caso presente, a um quadro de nefrite "mista", com lesões glomerulares de tipo glomerulonefrítico e alterações de estroma semelhantes às encontradas na nefrite intersticial e pielonefrite. Os ossos temporais foram removidos e foram processados. Lesões histológicas nestas estruturas foram descritas por Winter et co1.(7) em casos de nefrite hereditária com surdez. Outros achados de necrópsia foram cardiomegalia secundária a hipertensão e peritonite causada por E. coli e Pseudomonas.
DiscussãoO estudo clínico evolutivo dos membros da família afetados e o anatomopatológico de um dos elementos permite afirmarmos que se trata de nefropatia hereditária com surdez, principalmente pela presença das células espumosas ("Foam Cells")no rim. Estudos genéticos de Cohen e col.(8) sugerem quatro tipos de herança para esta moléstia: 1º - Dominância parcial ligada ao sexo. 2º -Dominância autossômica. 3º - Por: nefrotoxina transplacentária ou infecção uterina. 4º - Por segregação preferencial ou associação cromossômica. A transmissão genética não é fácil de explicar uma vez que elementos masculinos e femininos têm manifestação semelhante da moléstia, embora seja mais freqüentes em irmãos homens. Geralmente a moléstia se manifesta na segunda década da vida com nefropatia subclínica nos masculinos e surdez neurosensorial nas mulheres como afirma Turner(9). O pedigree da família em estudo demonstra a doença em antecedentes femininos e a predominância na quinta geração nos masculinos. Parcialmente ligada ao sexo (a localização do gene no cromossomo X e Y que são homólogos) foi sugerida na base do estudo de Perkoff e col.(10) em família Mórmon. Mas esta possibilidade está excluída por outras genealogias(11,12,13, 14, 15, 16, 17,18 19). Herança autossômica dominante com segregação anômala segundo Shaw, Glover(18) parece razoável. Mães heterozigotas transmitem o gene para mais da metade das filhas e provavelmente para mais de 50% aos filhos. Walker(l9) estudou a família (NP 666088) com pessoas afetadas em 4 gerações. Embora a histologia em dois casos estudados foi consistente com Síndrome de Alport incluindo a presença de células espumosas, ("Foam Cells") características atípicas incluindo falta de surdez em todas as pessoas afetadas inusualmente a sobrevida dos homens foi longa e a morte de uma mulher afetada ocorreu nos primeiros anos da segunda década da vida. As famílias estudadas por Ohlsson(l5) eram diferentes dos outros reportados porque miopia foi uma característica marcante e a disfunção renal nos homens afetados foi relativamente suave mesmo nos dois que tinham mais de 30 anos. A lesão renal se localiza primariamente no túbulo distal, na forma de glomérulonefrite difusa. A presença de ("Foam Cells") é patognomônico(20, 21, 22, 23) de síndrome hereditária. Em todos os membros afetados encontramos a hematúria e proteinúria de grau evolutivo. Krickstein, Gloor e Belogh(6) relatam que a doença apresenta dois aspectos interessantes: 1º - Nefrite hereditária com combinação entre glomerolonefrite, piolonefrite e nefrite intersticial e 2º - as ("Foam Cells") tem uma distribuição específica na córtex renal mais interna. Estes fatos confirmam os achados do caso em questão, como já o demonstrou o laudo do anatomopatologista. Todos os irmãos afetados com proteinúria e hematúria apresentavam disacusia neurosensorial. Os que eliminavam maior quantidade de proteínas tinham maior perda auditiva. As provas supraliminares mostraram sofrimento colear. Os estudos de Perkoff(24, 25, 26) mostram a queda em agudos no estudo audiométrico de seus pacientes. O estudo anatomopatológico do temporal não permitiu uma conclusão sobre as lesões cocleares devido a técnica histológica e a conservação do osso. Mas na cápsula labiríntica verificou-se a existência de um grande foco de otoesclerose sem fixação estapedial como vemos na fotografia nº 4. Não foram encontradas outras alterações na orelha média dos pacientes afetados com a doença. Nestes casos não acreditamos que os primeiros sintomas sejam a queda auditiva, zumbidos ou tonturas. Os pacientes primeiro descobrem o problema renal e após na investigação otológica aparece a lesão neurosensorial.

FIG. Nº 4 - Foco de otoesclerose na cápsula labiríntica, junto a platina estapedial no membro da família (Ciro). A platina estava móvel.
Estudamos uma família com vários membros portadores de surdez neurosensorial e nefropatia. Em todos a progressão da nefropatia piorava a audição. A queda neurosensorial em agudos estava presente em todos os casos com as provas supraliminares indicando sofrimento coclear. A presença de (" Foam Cells") no rim de um dos membros que faleceu mostrou que a família em estudo sofre de Síndrome de Alport ou nefropatia hereditária e surdez.
SummaryWe have studied a family in which several members presented sensory-neural hearing loss and nephropathy. In all of them the progressive kidney disease worsened the hearing loss. They presented susceptive deafness in the higher frequencies with all audiometric tests shouwing cocillear damage. The presente of foam cells in the kidneys of one of the patients made us conclude that this family presents Alport's syndrome or hereditary nephropaty and deafness.
Bibliografia1. Samelsohn, F. Über hereditare Nephritis und über den Hereditäts - Begriff in Allgemeinen. Vischows Arch. 59: 257, 1874.
2. Dickinxon, W. H., Diseases of the kidney and urinary derangements..Longmans Green 2: 379 1875.
3. Alport, A. C. Hereditary Familial eongenital hemorrhagic nephrits. Brit. Med. J. 1: 504, 1927.
4. Rinkoff, S. S. et al., Familial nephritis, report of cases and review of literature. Jama 113: 661, 1939.
5. Sohar, E. Renal disease, inner ear deaness, and ocular changes: new herede-familial syndrome. Ama Arch. Int. Med. 97: 627, 1956,
6. Krickstein, H. et al. Renal pathology in hereditary nephritis with nerve deafness. Arch Path, 82: 506, 1966.
7. Winter, L. E. et al. Hearing loss in hereditary renal disease. Arch. Otolaryngol, 88: 238, 1968.
8. Cohen, M. M., Cassady, G. and Hanna, B. L. "A genetic study of hereditary renal dysfunction with associated nerve deafness. Am. Hum. Genet. 13: 379-389, 1961.
9. Turner, J. S. Hereditary Hearing Loss with Nephropathy. Acta Oto. laryngol. supl. 271. 1970.
10. Perkoff, G. T. et al Clinical study of hereditary interstitial pyelonephritis. Ama. Arch. Int. Med. 88: 191, 1951.
11. Dockhorn, R. J. "Hereditary nephropathy without deafness. Am. J. Dia. Child. 114: 135-138, 1967.
12. Dubach, U. C. and Osell, D. R. "Alport's syndrome". Lancet 1: 159, 1961.
13. Marin, O. S. M. and Tyler, H. R. "Hereditary interstitial nephritis associated with polyneuropathy." Neurology 11: 999-1005, 1961.
14. Mulrow, P. J., Aron, A. M., Cathman, G. E:, Yesner, R. and Lubs, H. A. "Hereditary nephritis. Report of a Kindred. Am, J. Med., 35: 737-748, 1963.
15. Ohlsson, L. "Congenital renal disease, deafness and myopia in the family." Acta. Med. Scand. 174: 77-84, 1963.
16. Reyersbach, G. C., and Butler, A. M. "Congenital hereditary hematuria". New York J. Med. 251: 377-380, 1954.
17. Schneider, R. G., "Congenital hereditary nephritis with nerve deafness. New York J. Med. 63: 2644-2648, 1963.
18. Shaw, R. F. and Glover, R. A. " Abnormal secregation in hereditary renal disease with deafness. Am. J. Hum. Genet. 13: 89-97, 1961.
19. Walker, W. G. "Baltimore", Personal comunication, 1966.
20. Suzuki, Y. & Eanzaki, J. Alport's syndromo of familial kidney disease with deafness. J. Oto-Rhino-Laryng Soc Jap 67, 1175, 1964.
21. Chaptal, J. et al. Hereditary hematuric nephropathy with deafness (Alport's syndrome) remarks on two series of observations. Pediatrie 20, 649, 1965.
22. Case records of the Massachusetts general hospital: case 43511. New Engl. J, Med 257, 1231, 1957 (citado por J. S. Turner Jr.)
23. Goldbloom, R. & Haberfelde, G. C., Hereditary nephritis: report of a kindred. New Eng1 J Med 261, 734. 1959.
24. Perkoff, G. T. et al. Chronie hereditary nephritis and Y-chromosone linkage: reply to graham. Am J Hum Genet 12, 381. 1960.
25. Perkoff, G. T. Diseases of the kidney (ed. M. Strauss & L, Welt), pp. 953-960, Little, Brown, Boston-1963.
26. Perkoff, G. T. Familial aspects of diffuse renal diseases. Assoe Ren Med 15, 115, 1964.
Trabalho apresentado do XIV Congresso Argentino de Radiologia.
I Jornada Argentino-Brasileira de Radiologia - Paso De Los Libres (Corrientes) - Uruguaiana (Brasil) - 11, 12 e 13 de novembro de 1971.
* Trabalho executado no Instituto de Otologia de Porto Alegre.
** Médicos otologistas.
*** Médico geneticista.
**** Médicos residentes do Instituto de Otologia - Andradas, 1711, 3º andar.
***** Médica nefrologista (falecida).
****** Médico nefrologista.
