INTRODUÇÃOA hipoacusia congênita pode acompanhar síndromes caracterizadas por transtornos em vários órgãos e sistemas. Em alguns casos, a hipoacusia pode ser o primeiro sintoma de uma patologia ainda não descoberta, devendo o otologista estar alerta para a possibilidade de haver outros problemas associados com esta queixa7. Estima-se que cerca de 50% de todas hipoacusias neurossensoriais em crianças são devidos a fator genéticos, sendo a maioria transmitida por meio de gen recessivos2, 3, 7. Esta casuística é preocupante, visto que i maioria destes casos não existe tratamento eficaz. A prevenção então, torna-se o principal meio para reduzir sua eleva incidência7.
As hipoacusias genéticas podem ser congênitas ou início tardio, uni ou bilaterais, monossintomáticas ou sindrômica, As monossintomáticas são a maioria, correspondendo a 70% d disacusias neurossensoriais congênitas e sendo mais difíceis, serem diagnosticadas2, 7. A associação de síndromes genética com hipoacusia é muito freqüente7. O presente trabalho t por objetivo relatar três casos atendidos no Ambulatório Otologia do Hospital das Clínicas da Faculdade de Medicina Ribeirão Preto, da Universidade de São Paulo, e efetuar breve revisão da literatura pertinente.
Síndrome de Goldenhar
Vários termos têm sido usados para denominar e entidade: microssomia hemifacial, displasia oculoauriculovertebral, síndrome de Goldenhar-Gorlin, síndrome do primeiro e segundo arcos branquiais e displasia facial lateral. Aceita-se que o termo espectro óculo-aurículo-vertebral seja o mais correto4. É causada por herança multifatorial, apresentando história familiar positiva em 6% dos casos6.
É uma malformação complexa, predominantemente unilateral, que envolve estruturas derivadas do primeiro segundo arcos branquiais. A assimetria facial está presente, 65% dos casos, tornando-se mais aparente com o decorrer idade. Os ossos maxilar, temporal e malar, no lado mais intensamente comprometido, costumam ser reduzidos no tamanh em espessura. Em 10 a 30% dos pacientes o envolvimento bilateral, quase sempre exibindo um lado mais comprometido que o outro, sendo o mais freqüente o lado direito. Ocasionalmente, pode ainda ocorrer agenesia das glândulas salivares1, 4 Anormalidades oculares são comuns. O dermóide epibull ocorre em 35% dos casos. A blefaroptose e o estreitamento fissura palbebral ocorrem em 10% das pacientes. Anoftalmia e microftalmia têm sido reportadas em indivíduos c envolvimento severo, podendo estarem correlacionadas com retardo mental. A paresia do nervo facial está presente em 10% dos indivíduos, provavelmente causada por comprometime ósseo no nível do canal de Falópio1.
Estima-se que o déficit mental esteja presente em 5% a 15% dos indivíduos acometidos, podendo ser identificadas malformações do sistema nervoso central como: encefalocele, hidrocefalia, cisto dermóide, teratoma, malformação de Alnold-Chiari, cisto aracnóide e hipoplasia de corpo caloso1, 4.
Podem ser encontradas protuberâncias ou fístulas pré-auriculares como primeira manifestação. As anormalidades esqueléticas mais encontradas são de vértebras cervicais, como occipitalização do atlas, sinostoses e vértebras cuneiformes. Ocorrem ainda alterações estruturais do osso temporal que predispõem à meningite. Achados recentes relatam ainda associação com alterações cardíacas, como tetralogia de Fallot e defeito de septo ventricular; pulmonares, como: lobulação incompleta, hipoplasia ou agenesia pulmonar; renais, como: agenesia renal, duplo ureter, anormalidades vasculares, hidronefrose e hidroureter; sistema gastrintestinal, como: imperfuração anal com ou sem fístula retovaginal1, 4, 5.
Tanto perda auditiva condutiva, quanto neurossensorial têm sido reportadas em mais de 50% dos casos. A etiologia dessa perda é variável e pode significar anormalidades no ouvido médio e externo: hipoplasia da cadeia ossicular, nervo facial aberrante, anormalidades da tuba auditiva e de base do crânio4.
O prognóstico, obviamente, depende do fenótipo do paciente e da presença ou não de retardo mental4.
Síndrome de Stickler
Entidade de transmissão autossômica dominante com expressão variável, de ocorrência estimada em 1 para 20.000 pessoas, também conhecida por síndrome de Marshall-Stickler e síndrome de Wagner-Stickler ou artro-oftalmopatia hereditária. Face plana, fissura palatina, miopia intensa com descolamento de retina e catarata, perda auditiva e artropatia com displasia espondiloepifisária são suas características mais comuns. As manifestações craniofaciais podem variar da normalidade (15% a 25%), a alterações como: maxilar curto, olhos proeminentes, pregas epicantais, depressão do dorso nasal e queixo pequeno. Fenda palatina, anormalidades na mobilidade do palato têm sido reportadas em 20% dos casos1, 4, 9, 10. A miopia, que pode chegar a 18 dioptrias, é encontrada em 75% a 80% dos pacientes. Geralmente, é de início precoce, antes dos cinco anos de idade. Degeneração coriorretiniana e do vítreo ocorre antes dos 20 anos de idade em 70% dos acometidos e é freqüentemente bilateral. Se estas alterações não forem tratadas precocemente, podem levar à cegueira. Descolamento total da retina está presente em mais da metade dos pacientes, podendo ocorrer espontaneamente ou durante ato operatório, para correção de catarata. Outras alterações também encontradas são: astigmatismo (60%), catarata (45%), estrabismo (30%) e glaucoma (10%)4.
As articulações costumam ser ampliadas e freqüente mente, com hiperextensibilidade, algumas vezes sendo dolorosas e quentes. É descrita uma degeneração progressiva das articulações em 30% dos pacientes acometidos, parecendo não ser rara em indivíduos menores de 30 anos de idade. Prolapso de válvula mitral é encontrado em cerca de 50% dos casos4. Surdez neurossensorial costuma ser mais importante para altas freqüências e de caráter progressivo, estando presente em 80% dos pacientes com esta síndrome1, 4.
Síndrome de Marshall
Desordem de transmissão autossômica dominante de expressão variável. Miopia, catarata, nariz em sela, perda auditiva neurossensorial e hipertelorismo compõem as principais características desta síndrome. A falência da visão usualmente ocorre na segunda década de vida, havendo casos em que a perda visual ocorreu durante os primeiros seis meses, podendo a miopia ser maior que 10 dioptrias. São descritos ainda casos de descolamento de retina. Todos os pacientes apresentam severa depressão do dorso nasal e narinas antevertidas1, 4.
A perda auditiva neurossensorial costuma ser maior para altas freqüências e de início durante a infância. Muitas vezes pode ser progressiva e de aparecimento tardio, geralmente sem comprometimento vestibular1, 4.
Exames radiográficos podem evidenciar hipoplasia ou mesmo ausência dos ossos próprios do nariz, hipoplasia maxilar e ausência do seio frontal. São notadas ainda calcificações intracranianas, malformações vertebrais, pélvis irregular e pequena, retardo do fechamento dos ossos do púbis e ísquio, medialização da ulna e do rádio e, em alguns casos, irregularidade epifisária das extremidades4. Exame tomográfico de ossos temporais costuma revelar ouvidos internos normais e, em alguns indivíduos, estreitamento do canal auditivo interno. A falta do desenvolvimento do osso etmoidal pode ainda ocasionar o surgimento de uma fossa craniana anterior diminuída1.
O aconselhamento genético é útil na prevenção de novos casos. A cirurgia plástica pode trazer algum benefício para a correção do nariz em sela. A catarata pode ser corrigida pela cirurgia e a perda auditiva deve ser amenizada com aparelhos de amplificação sonora e uma educação especial1.
Caso 1 - Síndrome de Goldenhar
Paciente G. A. S., com 11 anos de idade, do sexo feminino, de cor branca, encaminhada ao Serviço de Otorrinolaringologia do Hospital das Clínicas, para avaliação auditiva, em seguimento na Genética Humana por assimetria de face desde o nascimento, e diagnóstico definitivo de Síndrome de Goldenhar. A mãe relata que a criança tem boa saúde, exceto por otalgias freqüentes e "ronqueira no peito", negando hipoacusia ou atraso no desenvolvimento mental ou da linguagem. Os pais não são consangüíneos, não havendo história de abordamento ou doença congênita na família.
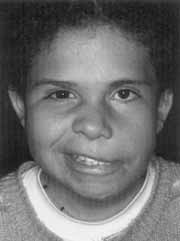
Figura 1. Síndrome de Goldenhar com paralisia facial periférica grau IV e assimetria facial (CASO 1).
Outro filho do casal goza de perfeita saúde física e mental. O exame físico revela face assimétrica, com hemiface esquerda hipodesenvolvida, região superciliar esquerda inferior em relação ao lado direito e microftalmia à esquerda. Nariz "em sela", com narina esquerda mais alargada. Rima bucal pouco desenvolvida, sendo desviada para a esquerda. Braquicefalia, osso frontal abaulado e com implantação baixa do couro cabeludo. Pescoço curto e com mobilidade normal. Hélix irregular, com chanfradura em sua região horizontal. Paralisia facial periférica grau IV de HouseBrackman5 à esquerda. Tórax de conformação normal, mamilos invertidos, genitália, períneo e coluna normais ao exame. Sindactilia de segundo e terceiro pododáctilos. Exame otoscópico revelou membranas timpânicas opacificadas e retraídas bilateralmente, com condutos auditivos normais. Orofaringoscopia mostra tonsilas palatinas grau II, sem hiperemia, secreções ou outra alteração. Exame audiométrico tonal liminar mostrou perda condutiva leve à direita em 6 e BKHz, e perda mista de grau moderado a severo à esquerda, com curva tipo B bilateral à imitanciometria. Exame oftamológico revelou coloboma de nervo óptico. Tomografia computadorizada de crânio constatou assimetria craniana em detrimento do lado esquerdo, sem outras alterações. Exame tomográfico de ossos temporais descartou malformação de ouvido interno e médio, evidenciando somente velamento de cavidade timpânica sugestiva de otite média crônica, sem qualquer erosão óssea. Raio X de cavum evidenciou hipertrofia adenoideana estreitando as vias aéreas; e exame radiográfico de coluna vertebral constatou espinha bífida em L4 e L5, com espaços discais conservados. Foi iniciada terapêutica clínica para otite média serosa com prednisolona, 0,5 mg por quilograma de peso corporal, por uma semana e beclometasona tópica nasal associado ao cetotifeno oral por três meses. Como a criança não obteve melhora após este período, foi indicada adenoidectomia, mais posicionamento de tubos de ventilação bilateralmente.
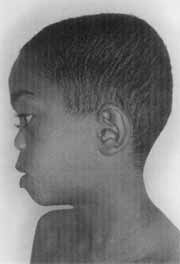
Figura 2. "Nariz em sela" e face plana característicos da Síndrome de Stickler (CASO 2).
Caso 2 - Síndrome de Stickler
Paciente E. F. D., com nove anos de idade, do sexo masculino, mulato, procedente de Pontal /SP, atendido inicialmente no Serviço de Cirurgia Pediátrica por criptorquidia e micropênis, sendo posteriormente encaminhado ao Serviço de Genética Humana, onde recebeu o diagnóstico definitivo de síndrome de Stickler. Foi solicitada avaliação otorrinolaringológica por suspeita de hipoacusia bilateral. Mãe relata que a criança ouve mal, assiste TV em volume muito alto, é desatenta e se irrita facilmente. Nega otites de repetição, respiração bucal de suplência ou roncos noturnos. Apresenta atraso do desenvolvimento da linguagem, emitindo somente poucos sons incovenientes.
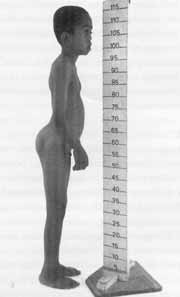
Figura 3. Face plana, baixa estatura, micropênis e artropatia características da Síndrome de Stickler (CASO 2).
Nega consangüinidade entre os pais, bem como casos de malformação na família. Exame otoscópico revelou membranas timpânicas íntegras pouco opacificadas, sem retrações, com condutos auditivos normais e implantação baixa de orelhas. Apresentava nariz em sela com septo centrado e cornetos sem alterações. Oroscopia evidenciou tonsilas palatinas grau II e dentes mal implantados. Exame ortopédico revelou ombros crepitantes à mobilização com acrômio saliente, cotovelos crepitantes e limitação da extensão, quadris com hipermobilidade em rotação, joelhos também crepitantes, principalmente à direita e com frouxidão ligamentar. Exame radiológico mostrou displasia epifisária em quadris, cotovelos e ombros, aumento do espaço articular escápulo-umeral, idade óssea compatível com quatro anos, pelo método de G. Pyle. Foi avaliado pela Oftalmologia, que diagnosticou hipermetropia, discreta proptose ocular e hipertelorismo. Audiometria tonal-liminar mostrou cofose bilateral e curva tipo C em imitanciometria e reflexo estapediano ausente. Exame tomográfico de ossos temporais revelou ainda malformação de ouvido interno tipo Mondini bilateral, cóclea com cavidade única e vestíbulo hipoplásico, hipoplasia de ouvidos médios com deformidade e espessamento de bigorna e estribo bilateralmente. Com este quadro, foi encaminhado ao setor de Fonoaudiologia para tentar adaptação de aparelho de amplificação sonora individual (AASI).
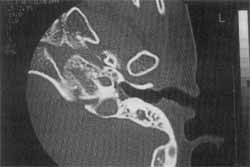
Figura 4. Tomografia de osso temporal esquerdo mostrando malformação de Mondini em paciente com Síndrome de Stickler (CASO 2).
Caso 3 - Síndrome de Marshall
Paciente R. V. D., com 4 anos de idade, do sexo masculino, cor branca, natural e procedente de Mirassol/ SP. Mãe refere que sua gravidez foi conturbada, tendo sido feito uso de hormônios (sic) na tentativa de aborto. Criança nasceu de parto cesariano, a termo. Pais não são consangüíneos e negam casos de malformações na família. Nega que a criança apresente hipoacusia ou atraso no desenvolvimento da linguagem. Foi atendido inicialmente pelo setor de Neurologia Infantil, por suspeita de macrocefalia. Exame revelou megalocórnea, prega epicântica, hipertelorismo, hipotonia global discreta, com força muscular preservada. Avaliação oftalmológica confirmou megalocórnea e miopia de alto grau (14,5 dioptrias à direita e 16 à esquerda). Foi encaminhado à Genética Humana, onde se confirmou o diagnóstico de síndrome de Marshall. Exame físico revelou hipoplasia acentuada de face média, raiz nasal baixa e fenda palpebral diminuída. O raio X de crânio mostrou textura óssea normal, desproporção crânio-facial, sem calcificações patológicas intracranianas. O exame otorrinolarigológico não revelou outras alterações além das descritas anteriormente, sendo solicitada audiometria condicionada, que mostrou audição dentro do padrão dê normalidade, imitanciometria com curva tipo A e reflexos presentes, sendo indicado retorno semestral para nova audiometria.
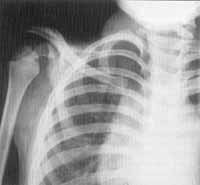
Figura 5. Displasia espondiloepifisária característica da Síndrome de Stickler (CASO .2).
Existem inúmeros tipos de hipoacusia genética, de modo que a probalidade de pessoas apresentarem o mesmo problema genético é muito pequena. 0 casamento entre pessoas com problemas auditivos não contribui necessariamente parra a maior incidência de hipoacusia hereditária, já que a chance de estes indivíduos serem portadores do mesmo defeito genético é mínima. Entre os casos por nós apresentados, nenhum apresentou história familiar positiva para doenças congênitas ou consangüinidade entre os pais, o que corrobora essa afirmação. Todavia, em algumas comunidades em que matrimônios consangüíneos são freqüentes, a incidência de hipoacusia genética, bem como de outras enfermidades hereditárias, é muito maior7
.
Em todos os casos, a avaliação por otorrinolaringologista foi solicitada, com a finalidade de descartar perda auditiva, ratificando a importância do otologista no seguimento dos pacientes com síndromes genéticas. No paciente com Síndrome de Goldenhar (Caso 1), os pais negavam que a criança apresentasse hipoacusia, tendo sido constatada perda mista de grau moderado a severo à esquerda e perda condutiva leve à direita, demonstrando a necessidade de uma boa avaliação audiométrica, mesmo quando esta queixa. é negada. Apesar de o exame tomográfico ter descartado malformação do ouvido interno, achamos que uma malformação do labirinto membranoso pode existir. Todavia, sua confirmação só seria possível por histopatologia, o que não é viável7.
Com relação ao paciente portador de síndrome de Stickler (Caso 2), o achado tomográfico de malformação tipo Mondini de ouvido interno, causando perda auditiva neurossensorial bilateral, assemelha-se aos descritos por Stickler (1976), restando tão somente a tentativa de amplificação sonora para amenizar seus efeitos sobre a criança, mesmo sabendo que uma adaptação pode ser muito difícil nestes casos.
No paciente portador da síndrome de Marshall (Caso 3), não foi constada perda auditiva, apresentando exame audiométrico tonal-liminar e imitanciométrico normais. A literatura alerta para casos em que a disacusia pode ser progressiva1, 4, o que aumenta a responsabilidade do otologista para continuar o seguimento do paciente com exames auditivos semestrais. Os demais achados referentes a estes pacientes foram semelhantes aos encontrados na literatura1 , 2, 3, 4, 6, 8.
As síndromes genéticas são entidades raras, que podem acometer o sistema auditivo, propiciando ser o otologista solicitado para efetuar avaliação minuciosa. Devemos, pois, estar aptos a identificar e diagnosticar patologias otorrinolaringológicas que eventualmente estejam associadas a doenças multissistêmicas. O conhecimento da causa da hipoacusia congênita facilitará o entendimento do sistema auditivo e o desenvolvimento da melhor abordagem terapêutica do paciente acometido pela enfermidade.
REFERÊNCIAS BIBLIOGRÁFICAS1. BUYSE, M. L.- Birth Defects Encyclopedia-A Seruice of the Center for Birth Defects Information Service, Inc., p.192-193; p.504-505, 1990.
2. DALLAPICCOLA, B; MINGARELLI, R.; GENNARELLI, M; NOVELLI, G.- Genetic aspects of deafness. Acta-OtorhinolaryngolItal., 16(2): 79-80, 1996.
3. ECKEL, H.E.; RICHLING, F.; STREPPEL, M.; ROTH, B.; WALGER, M.; ZOROWKA, P.- Etiology of moderade and profound deafness in childhood. HNO, 46(3): 252-263, 1998.
4. GORLIN, R. J.; TORIELLO, H. V.; COHENJR., M. M.- Hereditary Hearing Loss and Its Syndromes, Oxford University Press, p.6972, p. 120, p.193-194, New York, 1995.
5. HOUSE, J.W.- Facial nerve grading systems. Laryngoscope, 93: 1053-69, 1983.
6. MOCELLIN, M.; CAPASSO, R.; CATANI, G. S. A.; GASPERIN, A. C.; VIZZOTTO JR., A. O.- Síndrome de Goldenhar (Displasia Oculoauriculovertebral). Relato de caso e revisão de literatura. Rev. Bras. de Otorrinolaringol., 64(1): 77-79, 1998.
7. PAPARELLA, M. M.; SCHACHERN, P. A.- Hipoacusia neurossensorial genética en niños. In: Paparella, M.M; Shumrick, D. A.; Gluckman. J. L.; Meyerhoff, W. L.-.Enfermidades del oido, 3ª edição, 2: 1853-73, 1994.
8. SMITH, S.D.- Overview of genetic auditory syndromes- J. Am. Acad. Audiol., 6(1): 1-14, 1995.
9. STICKLER, G. B. E COLS.- Hereditary progressive arthro-ophthalmopathy. Mayo Clin. Proc. 40: 433-455, 1965.
10. STICKLER, G. B., PUGH, D. G. - Hereditary progressive arthro-ophthalmopathy: addicional observations on vertebral abnormalities, a hearing defect, and a report of a similar case. Mayo Clin. Proc. 42.- 495-500, 1967.
* Médico Residente em Otorrinolaringologia do Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto, da Universidade de São Paulo.
** Professor Doutor da Disciplina de Otorrinolaringologia do Departamento de Oftalmologia e Otorrinolaringologia da Faculdade de Medicina de Ribeirão Preto, da Universidade de São Paulo.
*** Professor Associado do Departamento de Genética e Matemática Aplicada á Biologia da Faculdade de Medicina de Ribeirão Preto, da Universidade de São Paulo.
Trabalho vinculado ao Departamento de Oftalmologia e Otorrinolaringologia da Faculdade de Medicina de Ribeirão Preto, da Universidade de São Paulo, e ao Departamento de Genética e Matemática Aplicada à Biologia da Faculdade de Medicina de Ribeirão Preto, da Universidade de São Paulo.
Apresentado no I Congresso Triológico de Otorrinolaringologia, entre 13 e 18 de novembro de 1999, em São Paulo/ SP.
Endereço para correspondência: Departamento de Oftalmologia e Otorrinolaringologia da Faculdade de Medicina de Ribeirão Preto, da Universidade de São Paulo.
Avenida Bandeirantes, 3.900 - 14049-900 Ribeirão Preto/ SP - Telefone: (0xx16) 633-1586.
Artigo recebido em 26 de janeiro de 2000. Artigo aceito em 11 de maio de 2000.
