INTRODUÇÃOA síndrome de Peutz Jeghers é relativamente rara; porém, bem conhecida e caracterizada por uma tríade: máculas melânicas na pele e mucosa, polipose gastrointestinal e tendência familiar. Alguns poucos trabalhos referem o comprometimento do trato aéreo superior, com o aparecimento de pólipos no seio maxilar e nasofaringel.
Esta síndrome foi descrita inicialmente em 19211, por Peutz, que mencionava em dois casos a presença de pólipos na nasofaringe. A síndrome foi reconhecida como entidade clínica distinta por Jeghers em 19491, quando descreveu dez casos de polipose gastrointestinal.
A pigmentação melânica é notada precocemente na infância; porém, pode aparecer mais tarde, com o passar dos anos. Acomete mais freqüentemente os lábios - 85% dos casos publicados, sendo menos comum nas extremidades e ao redor de outros orifícios faciais.
A transmissão da síndrome de Peutz Jeghers se faz de forma autossômica dominantes. Em alguns casos esporádicos, não se conseguiu evidenciar o caráter familiar`.
Os pólipos que surgem no trato digestivo podem ocorrer em qualquer lugar do mesmo, com predomínio no intestino delgado. Sob o ponto de vista anátomo-patológico, são benignos e do tipo harnartomatoso.
Apresentaremos a seguir dois casos atípicos da síndrome de Peutz Jeghers, pois achamos de valor de publicação dada a raridade da afecção e a concomitância com pólipo nasal.
CASO CLÍNICOCaso 1.
R. C. S., do sexo masculino, com 12 anos de idade, procedente de Guarulhos/SP, com história de obstrução nasal há um ano, sendo pior na fossa nasal direita, acompanhada de rinorréia grumosa e abaulamento da parede lateral direita do nariz. Há cinco meses, observou episódios de sangramento pela fossa nasal direita, que cessavam espontaneamente. Referia também cólicas abdominais freqüentes.
De antecedentes familiares, apresentava mãe com história de duas cirurgias do aparelho digestivo, devido a volvo (sic) há oito anos, além de manchas pigmentadas na mucosa oral, lábios e mãos.
Ao exame físico, apresentava abaulamento da parede látero-nasal direita, com presença de massa de superfície lisa ocupando toda a fossa nasal direita, além de secreção espessa, grumosa, de difícil remoção, no assoalho nasal. Observava-se ainda que esta massa desviava o septo nasal para a fossa nasal esquerda, ocluindo-a inteiramente. Além disso, notavam-se manchas hipercrômicas arredondadas ovaladas em lábios - semelhantes às de sua mãe (Foto 1) - bochechas e região cutânea perioral.
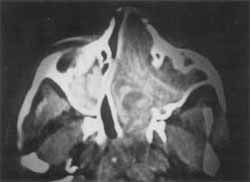
O exame tomográfico revelava massa ocupando as duas fossas nasais e o seio maxilar direito, empurrando o septo para o lado esquerdo, invadindo quase que totalmente o complexo maxilo-etmoidal (Foto 2). Não se individualizavam as estruturas de fossa nasal e havia lise óssea no nível da parede látero-nasal direita. A massa estendia-se posteriormente e ultrapassava o limite coanal, ocupando parcialmente a nasofaringe (Foto 3).
Fez-se o diagnóstico de síndrome de Peutz Jeghers, com pólipo gigante ocupando principalmente a cavidade nasal direita e seio maxilar direito. Foi indicada a ressecção cirúrgica por via transantral, com exposição pelo degloving. O tumor foi removido inteiramente; porém, como se tratava de massa friável, foi retirado em vários fragmentos. O muco que acompanhava o tumor era muito viscoso e de difícil aspiração. Ao final da cirurgia, o septo foi reposicionado para a linha mediana.
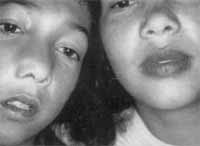
Foto 1.
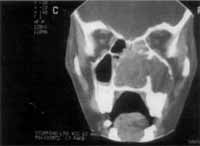
Foto 2.
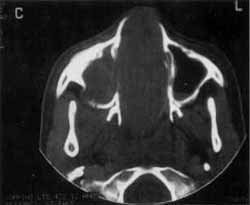
Foto 4.
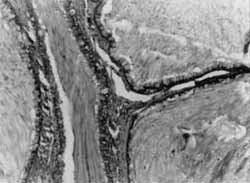
Foto 3.
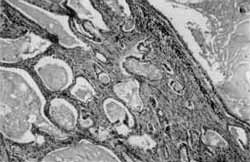
Foto 5.
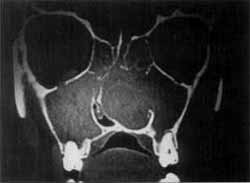
Foto 6.
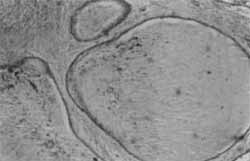
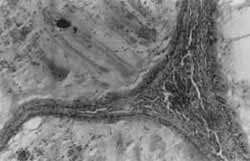
O exame anâtomo-patológico evidenciou pólipo hainartomatoso, com número aumentado de glândulas, algumas acentuadamente dilatadas (Foto 4), e estroma amplo, freqüentemente com fibras musculares lisas (Foto 5).
O paciente evoluiu bem, não apresentando recidiva da doença após dois anos de seguimento.
Caso 2.
A. P. L., do sexo feminino, com 12 anos de idade, estudante, natural de São Paulo/SP, com história de obstrução contínua em fossa nasal direita há seis meses, acompanhada de rinorréia amarelada e episódios de sangramento, além de abaulamento da parede lateral direita da pirâmide nasal. Referia também fortes cólicas abdominais freqüentes associadas à diarréia.
Ao exame físico, notava-se abaulamento da parede látero-nasal direita, observando-se, à rinoscopia anterior, uma massa de coloração avermelhada em fossa nasal direita, de consistência gelatinosa, indolor ao toque e pouco sangrante, que desviava o septo para a fossa nasal esquerda, além de grande quantidade de secreção espessa de difícil remoção. À rinoscopia posterior, observava-se que esta massa estendia-se para a rinofaringe. A paciente apresentava também manchas hipercrâmicas em lábio inferior e mucosa jugal.
O exame tomográfico evidenciava uma massa que ocupava a fossa nasal direita, empurrando o septo para a esquerda, com velamento dos seios maxilares e etmoidais (Foto 6) e que estendia-se para a rinofaringe (Foto 7).
Procedeu-se à cirurgia transantral, com exposição pelo degloving, com a retirada de toda a massa nasal, observando-se ainda erosão da parede anterior do seio maxilar direito, onde foi encontrada grande quantidade de secreção bastante espessa e grumosa, além de espessamento de sua mucosa, bem como a do seio etmoidal direito.
O exame anátomo-patológico evidenciou pólipo hamartomatoso, com inúmeras glândulas dilatadas (Foto 8), e estroma bastante amplo, com fibras musculares lisas (Foto 9).
A paciente evoluiu bem nos 30 dias iniciais de pósoperatório, quando, então, abandonou o acompanhamento clínico.
COMENTÁRIOSOs dois casos descritos são tipicamente de portadores de síndrome de Peutz Jeghers. Sabemos pela literatura que estes pacientes podem apresentar pólipo em qualquer parte do aparelho gastrointestinal, principalmente no intestino delgado - 64-96%d -, em concomitância com as manchas pigmentadas cutâneo-mucosas. No caso clínico 1, o paciente apresentava cólicas abdominais; e, segundo a mãe, estas eram muito semelhantes àquelas que ela sentia antes de ter sido operada do intestino há oito anos.
Segundo Bartholomew e cols., o ataque de cólicas severas abdominais é o sintoma mais freqüente, como apresentava a paciente do caso clínico 2, ocorrendo em 82% dos casos. Outros sintomas que podem ocorrer, além da dor, são: hemorragia digestiva, náuseas, melena e sinais de anemia. Existem aqueles que supõem que todos os casos apresentam polipose intestinal, embora a sua presença possa ser sub clínica.
Muitos autores mencionam a transformação maligna destes pólipos. Reide acredita que 2 a 3% dos pacientes adquirem câncer. Estes pólipos são hamartomas e geralmente não são considerados como lesão pré-maligna, embora alguns autores façam menção de uma grande incidência de lesões malignas gastrointestinais associadas a esses pólipos. Em decorrência desse fato, a malignidade poderia surgir como resultado da seqüência hamartoma-displasia-carcinoma ou surgir de tecido adjacente','°.
Existem trabalhos que referem a associação da síndrome de Peutz Jeghers com câncer de mama bilateral", tumor de ovário e adenoma maligno de útero` e tumores testiculares das células de Sertoli em crianças pré-puberais13.
A síndrome de PeutzJeghers tem caráter hereditário, sendo transmitida por um gene pleiotrópio autossômico dominante. Portanto, os pólipos e as manchas pigmentadas devem ser consideradas como uma manifestação pleiotrópica do mesmo defeito genético. A presença das manchas hipercrômicas na mucosa bucal e região perioral é decisiva para o diagnóstico".
A hiperpigmentação é plana, de cor parda escura ou parda azulada, de bordos irregulares e com diâmetro de 1 a 12 mm, sendo mais freqüentemente encontrada nos lábios, bochechas, palato duro e zona perioral, podendo ocorrer também ao redor dos olhos, nariz, extremidades de membros superiores e inferiores e regiões genital e periana114.
Histologicamente, as manchas na camada basal do epitélio epidérmico apresentam aumento de melanina e melanócitos. Estas lesões pigmentadas estão presentes muito cedo na infância, mas também podem desenvolver-se mais tarde. Aquelas da mucosa oral permanecem para sempre, enquanto que as das outras regiões podem desaparecer com o tempo'".
No caso 1, a mãe do paciente apresentava manchas na mucosa oral, lábios e também nos dedos das mãos, muito semelhantes às do filho. Desta forma, ficou evidente o caráter familiar.
Na síndrome de Peutz Jeghers, o aparecimento de pólipo no trato respiratório é raro, assim como no trato urogenitah. Após a primeira descrição de Peutz em 1921, poucos foram os casos publicados na literatura que fizessem referência ao comprometimento das vias aéreas superiores. Jancul, ao publicar em 1971 um caso desta síndrome onde, além do comprometimento do aparelho digestivo, o paciente também apresentava pólipo antromaxilar e de nasofaringe, referiu que Hartweg e Goerlich tinham relatado em 1960 um caso de paciente portador da síndrome de PeutzJeghers com pólipos no intestino e na cavidade nasal.
Mais recentemente, alguns outros poucos trabalhos também fizeram referência do envolvimento nasal na síndrome de Peutz Jeghers.
Conclue-se que a síndrome de Peutz Jeghers é uma entidade clínica rara, principalmente quanto a manifestações otorrinolaringológicas. A típica pigmentação dos lábios, face, dedos e mucosa oral que aparecem na infância, bem como polipose gastrointestinal, constituem as suas principais características, embora possam não estar presentes em todos os pacientes. Portanto, uma história clínica e familiar bem conduzida, além de exame físico cuidadoso, tornam-se muito importantes para que seja feito um diagnóstico preciso.
REFERÊNCIAS BIBLIOGRÁFICAS1. JANCU, J. - PeutzJeghers Syndrome. Amer. J. Gastroent., 56 545-49, 1971.
2. PEUTZ, J. L. A. - On a very remarkable case of familial polyposis of the mucous membrane of the intestinal and nasopharynx accompanied by peculiar pigmentations of the skin and mucous membrane. Ned. Tijdschr. Geneeskd., 10: 134-46, 1921.
3. JEGHER, H.; McCUSICK, V. A.; KATZ, K. H. - Generalized intestinal polyposis and melanin spots of the orai mucosa, lips and digists: a syndrome of diagnostic significance. New Engl. J. Med., 241: 993-1004, 1949.
4. BARTHOLOMEW, L. G.; MOORE, C. E.; WAUGH, J. M.Intestinal polyposis associated with mucocutaneous pigmentation. Surg. Gynec. Obstet., 115: 1-11, 1962.
5. DORMANDY, T. L. - Gastrointestinal polyposis with mucocutaneous pigmentation (PeutzJeghers Syndrome). New Engl. J Med., 256 1093-102, 1141-6, 1186-90, 1957.
6. KUWANO, H.; TAKANO, H.; SUGIMACHI, K. - Solitary PeutzJeghers type polip of the stomach in the absense of familial polyposis coli in a teenage boy. Endoscopy, 21: 188, 1989.
7. De FACA, L.; De SUTTER, J.; De MAN, M.; SPEK, P. V. D.; LEPOUTRE, L. - A case of PeutzJeghers Syndrome with Nasal Polyposis, Extreme Iron Deficiency Anemia, and Hamartoma-Adenoma Transformation: Management by Combined Surgical and Endoscopic Approach. Amer. J. Gastroent., 90: 1330-2, 1995.
8. McKITTRICK, J. E.; LEWIS, W. M.; GERWING, W. E. - The PeutzJeghers Syndrome. Arch. Surg., 103: 57, 1971.
9. REID, T. D. - Intestinal Carcinoma in the PeutzJeghers Syndrome. JAMA, 229.833-4, 1974.
10. SPIGELMAN, A. D.; MURDAY, V.; PHILLIPS, R. K. S. - Cancer and the PeutzJeghers syndrome. Gut., 30. 1588-90, 1989.
11. TRAU, H.; SCHEWACH-MILLET, M.; FISHER, B. K.; TSUR, H. - PeutzJeghers Syndrome and bilateral breast carcinoma. Cancer, 50: -788-92, 1982.
12. PODCZASKI, E.; KAMINSKI, P. F.; PEES, R C.; SINGAPURI, K.; SOROSKY, J. I.- PeutzJeghers Syndrome with ovarian sex cord tumor with annular tubules and cervical adenoma malignum. Gynecology Oncology, 42: 74-78, 1991.
13. DUBOIS, R. S.; HOFFMAN, W. H.; KRISHNAN, T. - Feminizing sex cord tumor annular tubules in a boy with PeutzJeghers. J Pediatr., 101: 568-71, 1982.
14. FERRARI, H. V. -Syndrome de PeutzJeghers (Presentación de un caso). Acta Gastroent. Latinoamer., 20 39-44, 199. 15. CERQUA, N.; D'OTTAVI, L. R.; PERROTTI, V. - Rare manifestations of nasal polyposis in the PeutzJeghers syndrome. Acta Otorhinol. Ital., 13: 333-8,1993.
* Doutorando do Curso de Pós-Graduação do Departamento de Otorrinolaringologia da Santa Casa de São Paulo.
** Chefe de Clínica do Departamento de Otorrinolaringologia da Santa Casa de São Paulo e Professor Adjunto de Otorrinolaringologia da Faculdade de Ciências Médicas de São Paulo.
*** Professor Assistente Doutor do Departamento de Ciências Patológicas da Santa Casa de São Paulo.
**** Residente do Terceiro Ano do Departamento de Otorrinolaringologia da Santa Casa de São Paulo. Pós-Graduando (Mestrado do Curso de Pós-Graduação do Departamento de Otorrinolaringologia da Santa Casa de São Paulo).
Trabalho realizado no Departamento de Otorrinolaringologia da Santa Casa de São Paulo. Trabalho apresentado, na forma de painel, no VI Congresso Brasileiro de Rinologia e Cirurgia Estética da Face e XIV International Symposium on Infectian and Allergy of the Nose (ISIAN), realizados em Salvador (BA), no período de 7 a 9 de Setembro de 1995. Endereço para correspondência: Rua Dr. Cesáreo Mota júnior, 112 - CEP: 01277-900 - São Paulo /SP. Artigo recebido em 26 de novembro de 1997. Artigo aceito em 16 de fevereiro de 1995.