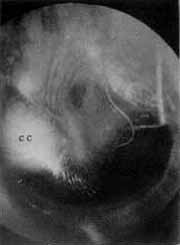
INTRODUÇÃOO colesteatoma congênito da orelha média é uma entidade clínica rara que, classicamente, apresenta-se como uma massa branca situada no quadrante ântero-superior da caixa timpânica, atrás de uma membrana timpânica intacta, associada ou não a uma perda auditiva condutiva ipsilateral 1. Esta afecção infreqüente pode também ocorrer em outras regiões do osso temporal: ápex petroso e conduto auditivo externo. Estima-se ainda que represente de 0,2 a 1,5% de todos os processos expansivos intracranianos, e cerca de 5 a 7% dos ângulos ponto - cerebelar (APC) 2 Acredita-se que se origine de remanescentes epiteliais aberrantes, mostrando preferência por certas localizações intracranianas. Estes sítios prediletos incluem: o APC, região do quiasma óptico, díploe do crânio e fossa rombóide 3. VALVASORI 4 classificou as formações epidermóides do osso temporal em 4 localizações: caixa do tímpano/mastóide, pirâmide petrosa, APC e fossa jugular (Figura 1).
Entre nós, BARRIONUEVO e col. em 1974 5 e CAMPANA e col. em 1988 6 descreveram casos pessoais de colesteatomas congênitos.
Atribui-se a DUVERNEY a primeira descrição de um "tumor epidermóide " intracraniano, feita em 1683, que o mesmo chamou de "esteatoma". CRUVEILHIER', em 1829, referiu - se a lesões similares como "tumores perláceos". PERON e SCHUKNECHT 8 relatam que MÜLLER, em 1938, foi o primeiro a usar o termo "colesteatoma" para descrever depósitos intracranianos de queratina.
EGGSTON e WOLFF 9 relatam que a teoria de que os colesteatomas originam-se de restos epiteliais, surgindo como "tumores epidermóides" no transcorrer da vida, foi apresentada inicialmente por KORNER e VIRCHOW (1830) e apoiada por CUSHING e MacKENZIE na primeira metade do século XX. Entretanto, a clássica descrição de um colesteatoma da orelha média atrás de uma membrana timpânica íntegra só foi relatada inicialmente por HOUSE em 1953, como referem LASKIEWICZ et al 10.
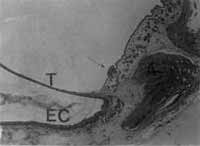
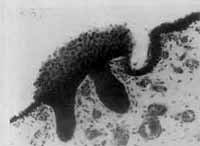
Diversos mecanismos têm sido propostos para explicar a origem do colesteatoma congênito. A noção mais habitualmente aceita de restos epidermóides, proposta por TEED 11 em 1936, sugere que os colesteatomas congênitos se originariam de restos ectodérmicos que se desenvolvem a partir das primeiras estruturas epibranquiais dos peixes e aves. Aberrantes tecidos ectodérmicos foram descritos no epitímpano anterior de embriões humanos'. Mais recentemente, MICHAELS 12 (1986) mostrou em estudos histológicos de fetos humanos, uma formação epidermóide na porção ântero-superior da mucosa da orelha média. Ela se distribui uniformemente próximo à origem da membrana timpânica e adjacente ao bordo posterior do anel timpânico, desaparecendo por volta da 33a semana de gestação, em fetos normais, podendo, no entanto, persistir e ter o potencial de se desenvolver num colesteatoma congênito. Recentes estudos de LEVENSON et al 13 (1986) e COHEN' 14 (1987) mostram que dois terços dos colesteatomas congênitos originam-se no quadrante ântero-superior da orelha média, o que falaria a favor desta teoria. Esta formação epidermóide também foi observada por WANG et al 15 (1987), porém numa menor incidência. Outros autores, como SCHUKNECHT 16 (1988), acreditam que a formação epidermóide é muito mais anterior (i. e, anterior à membrana timpânica) para dar origem a colesteatomas congênitos (Figuras 2 e 3).
AIMP 7 sugere a hipótese de que, quando o primitivo anel timpânico falha em prover o seu sinal usual que controla o crescimento interno da pele da parede do conduto auditivo externo, haveria a possibilidade de que projeções ectodérmicas do conduto em desenvolvimento passariam, através do istmo timpânico, para dentro do mesênquima epitimpânico, gerando um foco para a formação de um cisto epidermóide.
DESA 18, em 1983, propôs a seguinte teoria: mesênquima embrionário, normalmente presente na fenda da orelha média de neonatos, habitualmente sofre um processo de reabsorção que dura cerca de 1 ano. Levando-se em consideração que este tecido é pluripotente, haveria a possibilidade do mesmo sofrer um processo de diferenciação, transformando-se em epitélio escamoso queratinizado e, desta forma, num colesteatoma congênito.
SADÉ et al 2 (1983) propuseram a teoria da metaplasia escamosa da mucosa da orelha média, enquanto que PAPARELLA e RYBAH 9 (1978) a da origem a partir de tumores epidermóides localizados lateralmente na cavidade timpânica e que se formam a partir da fusão de planos do primeiro e segundo arcos branquiais. Em 1985, PIZA et al 20 aventaram a hipótese de que restos escamosos presentes no líquido amniótico poderiam desenvolver-se num colesteatoma congênito.
Existem estudos mostrando que a incidência dessa doença obedece a uma relação de 3 homens para 1 mulher 21,22 JAFEK et al 23 (1975) notaram uma preponderância masculina de 65% numa extensa revisão de atresia aural congênita, que foi considerada estatisticamente significante.
CASOS CLÍNICOS1) C. A., 12 anos, sexo feminino.
HPMA: diminuição da audição no ouvido direito; a otoscopia sugeria um quadro de otite média secretora; a miringotomia revelou tecido epidermóide na orelha média.
DESCRIÇÃO CIRÚRGICA: colesteatoma tomando a caixa timpânica, ático e antro; estribo e bigorna destruídos. Realizada timpanoplastia com mastoidectomia, com ampla abertura do recesso do facial. Feita interposição da bigorna moldada entre martelo e platina do estribo.
AUDIOMETRIA: (média das freqüências 0,5, 1 e 2 kHz)
VIA ÓSSEA PRÉ: 10 dB VIA AÉREA PRÉ: 55 dB
GAP PRÉ: 45 dB
VIA ÓSSEA PÓS: 10 dB VIA AÉREA PÓS: 45 dB
GAP PÓS: 35 dB
GANHO 10 dB
RESULTADO FUNCIONAL: GANHO AUDITIVO.
RESULTADO ANATÔMICO: BOM
Seguimento: Controle após 8 anos: enxerto bem; porém retraído.
2) R. M., 3 anos, sexo masculino.
HPMA: dor no ouvido direito freqüente; otoscopia revelou pérola branca na orelha média.
DESCRIÇÃO CIRÚRGICA: colesteatoma na caixa timpânica, com envolvimento de toda a cadeia ossicular. Realizada timpanoplastia com mastoidectomia, onde se observou granuloma de colesterol na mastóide. Não houve certeza da retirada completa do colesteatoma. Não foi feita nenhuma reconstrução funcional.
AUDIOMETRIA:
VIA ÓSSEA PRÉ: 10 dB VIA AÉREA PRÉ: 35 dB
GAP PRÉ: 25 dB
VIA ÓSSEA PÓS: 25 dB VIA AÉREA PÓS: 55 dB
GAP PÓS: 30 dB
GANHO: -5 dB
RESULTADO FUNCIONAL: SEM GANHO AUDITIVO
RESULTADO ANATÔMICO: BOM
Seguimento: mastoidite aguda 2 anos após a cirurgia; recidiva do colesteatoma; realizada mastoidectomia radical.
3) A. A. T., 12 anos, sexo masculino.
HPMA: supuração com sangue há 2 dias pelo ouvido esquerdo. Já escutava pouco. A otoscopia revelou pérola na região ântero-superior da membrana timpânica.
DESCRIÇÃO CIRÚRGICA: colesteatoma na caixa timpânica e "aditus". Granuloma de colesterol na mastóide. Realizada timpanoplastia com mastoidectomia. A bigorna e estribo estavam destruídos. Não fizemos reconstrução da cadeia ossicular.
AUDIOMETRIA:
VIA ÓSSEA PRÉ: 5 dB VIA AÉREA PRÉ: 55 dB
GAP PRÉ: 50 dB
VIA ÓSSEA PÓS: 25 dB VIA AÉREA PÓS: 75 dB
GAP PÓS: 50 dB
GANHO: 0 dB
RESULTADO FUNCIONAL: PIORA DA AUDIÇÃO (SENSORIONEURAL).
RESULTADO ANATÔMICO: MAU
Seguimento: mastoidite aguda 20 meses após a cirurgia. Recidiva do colesteatoma; realizada mastoidectomia radical.
4) C. P., 6 anos, sexo feminino.
HPMA: apresentou um episódio de otite média aguda à esquerda com paralisia facial periférica. A miringotomia revelou massa epidermóide na caixa timpânica.
DESCRIÇÃO CIRÚRGICA: pérola de colesteatoma na fenda da orelha média, envolvendo a cadeia ossicular; erosão do ramo longo da bigorna; presença de granuloma de colesterol na mastóide: Realizada timpanoplastia com mastoidectomia. Interposição da bigorna moldada entre martelo e platina do estribo.
AUDIOMETRIA:
VIA ÓSSEA PRÉ: 10 dB VIA AÉREA PRÉ: 35 dB
GAP PRÉ: 25 dB
VIA ÓSSEA PÓS: 10 dB VIA AÉREA PÓS: 40 dB
GAP PÓS: 30 dB
GANHO: -5 dB
RESULTADO FUNCIONAL: SEM GANHO AUDITIVO
RESULTADO ANATÔMICO: BOM
Seguimento: membrana timpânica um pouco retraída após 3 anos e meio.
5) L. P. F. N., 8 anos, sexo masculino.
HPMA: removeu um pólipo do ouvido direito há dois anos e desde então apresenta supuração fétida. Nunca teve problemas de ouvido antes.
DESCRIÇÃO CIRÚRGICA: enorme colesteatoma tomando toda a caixa da orelha média e mastóide, com exposição de meninge. Todos os ossículos destruídos. Realizada timpanoplastia com mastoidectomia. Não foi feita ossiculoplastia.
AUDIOMETRIA:
VIA ÓSSEA PRÉ: 10 dB VIA AÉREA PRÉ: 20 dB
GAP PRÉ: 10 dB
VIA ÓSSEA PÓS: 10 dB VIA AÉREA PÓS: 20 dB
GAP PÓS: 10 dB
GANHO: 0
RESULTADO FUNCIONAL: AUDIÇÃO INALTERADA
RESULTADO ANATÔMICO: BOM
Seguimento: revisão da cirurgia após 2 anos e 4 meses: presença de colesteatoma residual. Realizada mastoidectomia radical.
6) O. H. Y., 8 anos, sexo masculino.
HPMA: agenesia congênita do ouvido esquerdo, referindo escutar menos do ouvido direito; sem supuração. A otoscopia evidenciou pérola branca na orelha média direita atrás de membrana timpânica íntegra.
DESCRIÇÃO CIRÚRGICA: grande colesteatoma tomando toda a caixa timpânica e mastóide. Todos os ossículos destruídos. Realizada timpanoplastia com mastoidectomia. Não foi feita ossiculoplastia.
AUDIOMETRIA:
VIA ÓSSEA PRÉ: 15 dB VIA AÉREA PRÉ: 60 dB
GAP PRÉ: 45 dB
VIA ÓSSEA PÓS: 15 dB VIA AÉREA PÓS: 55 dB
GAP PÓS: 40 dB
GANHO: 5 dB
RESULTADO FUNCIONAL: AUDIÇÃO INALTERADA
RESULTADO ANATÔMICO: BOM
Seguimento: revisão da cirurgia após 2 anos e meio: presença de colesteatoma residual. Realizada mastoidectomia radical modificada.
7) A. M. R., 21 anos, sexo feminino.
HPMA: diminuição da audição à direita desde criança e há 18 meses notou diminuição dos movimentos da face do mesmo lado. Nega supuração ou vertigem. A otoscopia mostrou enorme massa branca ocupando a fenda da orelha média e membrana timpânica íntegra. O estudo tomográfico mostrava uma grande massa ocupando toda a fenda da orelha média que invadia a orelha interna com destruição de cóclea e canais semicirculares (figura 4 e 5).
DESCRIÇÃO CIRÚRGICA: grande colesteatoma ocupando caixa timpânica e mastóide, com destruição da cóclea, canais semicirculares lateral e posterior e exposição de meninge
e de todo o facial na sua porção horizontal. Todos ossículos comprometidos. Realizada mastoidectomia radical modificada sem ossiculoplastia, labirintectomia e descompressão do nervo facial.
AUDIOMETRIA: ANACUSIA À DIREITA. RESULTADO ANATÔMICO: BOM
Seguimento: evolução boa após 1 mês de cirurgia.
DISCUSSÃONo passado, colesteatomas congênitos limitados á orelha média eram raramente diagnosticados; isto fica evidente quando, em 1935, TEED 11 , revendo a literatura até então, encontrou apenas 5 casos relatados. Provavelmente este fato deve-se à ausência de parâmetros definidos para um diagnóstico preciso da doença, o que só ocorreu em 1953, quando HOUSE publicou o primeiro caso de um colesteatoma atrás de uma membrana timpânica íntegra. Após isto, o número de casos de colesteatomas congênitos vem aumentando exponencialmente, tanto que no período de 1975 a 1989 120 novos casos da doença foram publicados 3.
O colesteatoma congênito da orelha média usualmente origina-se no mesotímpano anterior e, com o passar do tempo, estende-se em direção aos ossículos no mesotímpano posterior e, com igual freqüência, ao epitímpano anterior. Quando pequeno, consiste habitualmente de um cisto queratótico unilocular, circulando por mucosa normal, conferindo uma aparência perlácea ao segmento ântero-superior da membrana timpânica. Com o posterior crescimento, ele pode estender-se ao orifício da tuba auditiva, levando a um quadro de otite média secretora, ou erosar a cadeia ossicular, principalmente a articulação incudoestapedial, resultando em perda auditiva condutiva.
PERON e SCHUKNECHT8(1975) publicaram o primeiro estudo histopatológico do osso temporal de um paciente com colesteatoma congênito da orelha média, que serviu de base para que McGILL et al 1 definissem dois tipos distintos: a) tipo "fechado": cisto queratótico, localizado atrás do quadrante ântero-superior da pars tensa. Este colesteatoma está presente ao nascimento e cresce lentamente para, na infância, apresentar - se como uma massa branca atrás de uma membrana timpânica íntegra, sendo habitualmente assintomático ou levando algumas vezes à deficiência auditiva condutiva. Quando pequeno, é facilmente removido na sua totalidade através de uma timpanotomia exploradora. A maior parte dos casos vistos na prática clínica encaixa-se nesta categoria; b) tipo "aberto": a matriz do colesteatoma cobre parte da mucosa da orelha média - usualmente a metade anterior da pars tensa. A matriz pode estender-se e recobrir outras estruturas da orelha média, como ossículos e músculos intratimpânicos. Esta variedade, clinicamente rara, necessita de um tratamento cirúrgico mais amplo, com a retirada de porções da membrana timpânica, ossículos; músculos e toda a mucosa recoberta pela matriz, para impedir eventuais recidivas.
De todas as teorias que foram propostas para explicar a patogênese dos colesteatomas congênitos, a que mais se apóia em evidências científicas é a que cita a existência de uma formação epidermóide embriológica na orelha média, que persistiria após o nascimento, terminando por desenvolver-se num colesteatoma.
TEED 11 (1936) publicou um intrigante estudo intitulado "Cholesteatoma Verum Tympani", no qual descreve uma estrutura epidermóide em um feto humano de 5 meses e meio, na porção dorsolateral da membrana timpânica, justamedial ao colo do martelo. Ele concluiu que "células epidermóides estão normalmente presentes tio pólo dorsolateral do tímpano e que as mesmas transformam-se normalmente em células epiteliais. Ocasionalmente, entretanto, elas persistem na sua qualidade ectodérmica e produzem pele, que, descamando-se, origina o colesteatoma". Os achados de TEED ficaram esquecidos por quase 50 anos quando, em 1986, MICHAELS 12, revendo os ossos temporais de fetos humanos, redescobriu uma estrutura epidermóide que era identificável da 10a à 33a semana de gestação, em 37 das 68 orelhas estudadas. Esta formação epidermóide era encontrada na mucosa ântero-superior da orelha média, próxima à origem da membrana timpânica e adjacente ao bordo posterior do osso timpânico. Estudos posteriores confirmaram que esta formação epidermóide é uma estrutura embriológica normalmente encontrada, desempenhando, talvez, o papel de organizadora do desenvolvimento da membrana timpânica e da orelha média. Tal estrutura situa-se numa região de transformação histológica em que se observa a transição do epitéliocuboidal simples, que reveste a cavidade timpânica na sua porção posterior, em epitélio pseudo - estratificado, freqüentemente ciliado, presente na cavidade timpânica anterior e tuba auditiva.
A formação epidermóide parece desempenhar uma ativa função no desenvolvimento da mucosa da orelha média a partir da tuba auditiva, e de organizadora do desenvolvimento da cavidade timpânica, tracionando, no sentido posterior, o endoderma da bolsa faríngea em direção ao endoderma da fenda branquial. Este processo se completa por volta da 33a semana de gestação, após a qual observa-se uma involução natural desta formação.
Parece razoável acreditar que o colesteatoma congênito da orelha média resultaria de uma falha na involução normal da formação epidermóide. Como resultado, teríamos uma
proliferação contínua de suas células. É interessante notar que, se a formação epidermóide continuar a expandir-se, seria de se esperar o seu aparecimento no quadrante ântero-superior da orelha média, o que é comprovado na prática clínica, pois é esta a região onde mais freqüentemente se observam os colesteatomas congênitos.
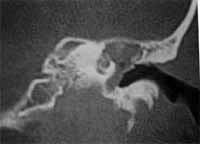
Figura 4
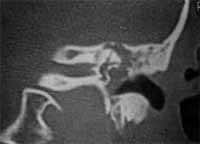
Figura 5
Embora o colesteatoma congênito seja uma entidade clínica rara, sua ocorrência não pode passar despercebida. O excelente prognóstico,, ainda com a lesão confinada ao quadrante ântero - superior, faz do diagnóstico precoce uma necessidade absoluta. O fato de saber-se que, em casos onde o colesteatoma congênito encontra-se em fase inicial, uma simples timpanotomia exploradora pode ser suficiente para erradicar a doença, deveria encorajar o emprego de medidas que permitissem o seu diagnóstico o mais cedo possível.
Embora de comportamento mais benigno que o do colesteatoma adquirido, os casos relatados mostram que o diagnóstico feito em fases avançadas da doença leva o otologista
a realizar cirurgias extensas, com comprometimento do resultado funcional.
Deve ser difundida a importância do diagnóstico precoce, a fim de impedir o crescimento silencioso desta doença, evitando a necessidade de cirurgias amplas e a ocorrência de destruições extensas e seqüelas irreversíveis, como as do caso clínico sete.
AGRADECIMENTOOs autores agradecem ao Dr. Ivo Bussoloti Filho pela documentação fotográfica apresentada neste trabalho.
REFERÊNCIA BIBLIOGRÁFICA1. McGILL, T. J.; MERCHANT, S.; HEALY, G. B.; FRIEDMAN, E. M. - Congenital cholesteatoma of the middle ear in children: a clinical and histopathological report. Laryagoscope, 101: 606 - 13, 1991.
2. SADÉ, J.; BABIACK, A.; PINKUS, G. -The metaplastic and congenital origin of cholesteatoma. Acta Otolaryngol, 96: 119 - 29, 1983.
3. CHEN, J. P.; SCHLOSS, M. D.; MANOUKIAN, J. P.; SHAPIRO, R. S. - Congenital cholesteatoma of the middle ear in children. J. Otolaryngol,18 (1): 44 - 8, 1989.
4. VALVASSORI, G. E. - Benign tumors of the temporal bone. Radiol Clin North Am, 12: 533 - 42, 1974.
5. BARRIONUEVO, C. E.; CIDRAL, W.; WITTIG, E. - O Colesteatoma Congênito, Rev. Brasil. 0. R. L., 40 (2-3):302 - 303, 1094.
6. CAMPANA, D. R.; SALEM, L.; PALERMO, F. R.; SARMENTO NETO, P. M.- O Colesteatoma Congênito. Apresentação de 3 casos. Rev. Brasil. 0. R. L., 54 (4):119-122,1988.
7. CRUVEILHIER, L. J. B. - Anatomie pathologique du corps humain. Livre 1 - 20, Bailliere, Paris, 1844, pp 1829 - 35.
8. PERON, D. L.; SCHUKNECHT, H. F. - Congenital cholesteatoma with other anomalies. Arch Otolaryngol, 101: 498 - 505, 1975.
9. EGGSTON, A.; WOLFF, D. - Histopathology of the Ear, Nose and Throat. Willians & Wilkins Co., Baltimore, p. 432, 1947.
10. LASKIEWICZ, B.; CHALSTREY, S.; GATLAND, D. J.; JONES, N.; MICHAELS, L.; PATH, F. R. C. - Congenital cholesteatoma. J. LaryngolOtol, 105.995 - 8, 1991.
11. TEED, R. W. -Cholesteatomaverumtympani: its relationship to the first epibranchial placode. Arch Otolaryngol, 24 455 - 74,1936.
12. MICHAELS, I. -An epidermoid formation on the developing middle ear: possible source of cholesteatoma. J. Otolaryngol, 15: 169 - 74, 1986.
13. LEVENSON, M. J.; PARISIER, S. C.; CHUTE, P.; WENIG, S.; JUARBE, C. - A review of 20 congenital cholesteatomas of the middle ear in Child ren. Otolaryngol Head and Neck Surg, 94. 560 - 7, 1986.
14. COHEN, D. - Location of primary cholesteatoma. Am J. Otol 8.61 - 5, 1987.
15. WANG, R. G.; HAWKE, M.; KWOK, P. - The epidermoid formation (Michaels'structure) in developing middle ear. J. Otolaryngol,16: 327 - 30, 1987.
16. SCHUKNECHT, H. F. - Epidermoid formation in fetal ears. Correspondence. J. Otolatyngol, 17: 62, 1988.
17. AIMI, K. - Role of the tympanic ring in the pathogenesis of congenital cholesteatoma. Laryngoscope, 93: 1140 - 6, 1988.
18. DESA, D. J. - Mucosal metaplasia and chronic inflammation in the middle ear of infants receiving intensive care in the neonatal period. Arh Dis Child, 58:24 - 8, 1983.
19. PAPARELLA, M. M.; RYBAH, L.-Congenitalcholesteatoma. Otolaryngol Clin North Am, 11: 113 - 20, 1978.
20. PIZA, J.; NORTHOP, E. C.; EAVEY, R. - Amniotic fluid contents in the ears of neonates and infants. XIII World Congress of Otorhinolaryngology (Abstract), 1: 48, 1985.
21. SANNA, M.; ZINI, C. - Congenital cholesteatoma of the middle ear: a report of 11 cases. Am J. Otol., 5:368 - 78, 1984.
22. SCHWARTZ, R. H.; GRUNDFAST, K. M.; FELDMAN, B. et ai - Cholesteatoma medial to an intact tympanic membrane in 34 young children. Pediatrics, 74: 236 - 40, 1984.
23. JAFEK, B. W.; NAGER, G. T.; STRIFE, J. et ai - Congenital aural atresia: an analysis of 311 cases. TransAm Acad Opth Otol, 80: 588 - 95, 1975.
24. CAWTHORNE, T. - Congenital cholesteatoma. Arch Otol, 78: 248 - 52, 1963.
Trabalho do Departamento de Otorrinolaringologia da Santa Casa de Misericórdia de S. Paulo e Disciplina de Otorrinolaringologia do Departamento de Oftalmo e Otorrinolaringologia da Fac. Ciências Médicas da Santa Casa de S. Paulo.
* Aluno do Curso de Pós - Graduação em Otorrinolaringologia, da Fac. Ciências Médicas da Santa Casa de S. Paulo.
** Diretor do Departamento de Otorrinolaringologia da Santa Casa de Misericórdia de S. Paulo e Titular de Otorrinolaringologia da Fac. Ciências Médicas da Santa Casa de S. Paulo. Coordenador dos Cursos de pós-graduação em Otorrinolaringologia.
*** Chefe de Clínica Adjunto do Departamento de Otorrinolaringologia da Santa Casa de Misericórdia de S. Paulo e Professor Assistente de Otorrinolaringologia da Fac. De Ciências Médicas da Santa Casa de S. Paulo.
Endereço dos autores: Departamento de Otorrinolaringologia da Santa Casa de I S. Paulo - Rua Dr. Cesario Motta Sr., 112,4Q andar - CEP 01227-000 - São (Paulo - SP.
Artigo recebido em 24 de janeiro de 1994. (Artigo aceito em 24 de fevereiro de 1994.