São pouquíssimas as informações existentes, na literatura médica, sobre a associação entre imperfuração coenal e outras malformações congénitas(GRAHNE e KALTIOKALLIO, 1966). Por isso, achamos interessante divulgar um caso de oclusão coanal congénita unilateral, por nós operado recentemente, num paciente portador da Síndrome de Apert (ACROCEFALO-SINDACTILIA). Nenhuma citação desta associação foi encontrada na bibliografia consultada.
RELATO SUMARIO DO CASO0 menor R.C.M., de 16 anos, branco, do sexo masculino, brasileiro, nos foi trazido por sua mãe com a queixa principal de má respiração nasal crônica. Por se tratar de um retardado mental, incapaz de responder às perguntas formuladas, todas as informações, pertinentes ao caso, foram obtidas através de sua própria mãe e das observações contidas em seu prontuário médico do H.C.Aer., hospital onde nasceu e foi repetidas vezes internado para tratamentos vários. Contamos também com as informações do Dr. Cyldes da Silva, pediatra que o assistiu desde o nascimento até quase a data atual, tendo sido o primeiro a firmar o diagnóstico de Síndrome de Apert.
Dado o grande número de anormalidades existentes, apenas registramos as mais significativas.
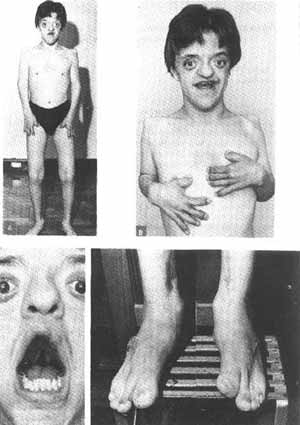
Nascido a termo, mas extraído a forceps, o paciente apresentava acrocefalia (operou com sucesso, quando lactente, as suturas a fim de permitir a expansão do encéfalo), hipertelorismo, exoftalmia, rínomegalia, hipoplasia do maxilar superior e prognatismo do maxilar inferior, dentes mal implantados, fenda palatina (operada várias vezes, mas com sucesso parcial), sindactilia completa de ambos os pés e mãos (as mãos foram operadas com sucesso; em ambos os pés, apenas foram isolados o primeiro do segundo pododactilo), soldadura prococe das cartilagens de conjugação dos membros superiores, malformações articulares dos ombros, cotovelos, pés e mãos, desvio do septo para direita, malformação das fissuras orbitárias e arcadas orbitárias superiores, e hérnia inguinal congênita (operada com sucesso). A maior parte das malformações citadas são facilmente identificadas nas figuras A, B, C e D.
0 exame O.R.L. atual confirmou a presença de uma deformidade septal grau III (implantação) ao nível das áreas 2,4 e 5 de Cottle (áreas da válvula, dos cornetos e da coana), bloqueando praticamente toda a fossa nasal direita, e abundante secreção catarral. Não nos foi possível realizar exame mais pormenorizado, incluindo rinoscopia posterior, rinomanometria, audiometria ou mesmo simples testes com diapasões, por absoluta falta de entendimento e colaboração do enfermo. Um estudo radiológico revelou-nos seios da face de tamanho, simetria e transparência normais.
Todavia, plenamente justificada a obstrução nasal crônica pela presença de deformidade septal, foi o paciente levado à cirurgia em 08 de junho de 1976. Foi feita uma reconstrução septal do tipo 3, (segundo a classificação de Guillen-Neves Pinto (NEVES PINTO, 1975), via maxila-premaxila de COTTLE Col. (1958), sob anestesia geral. Ao investigarmos a área 5 do septo nasal, deparamos com um diafragma ósseo que ocluia completamente a coana direita. Foi então removido com pinças ósseas, através do espaço intrasseptal, segundo a mesma técnica descrita em trabalho recente (NEVES PINTO e DE BONA, 1976). Infelizmente, não nos foi possível documentar a imperfuração coaria(, assim constatada durante a cirurgia septal, por meio de rediografia contrastada, pela falta de equipamento disponível naquele exato momento. 0 paciente evoluiu bem. Apesar de não permitir a inspecção demorada das fossas nasais (não tolera o uso do especulo), surpreendentemente, não criou o menor problema para o tamponamento nasal que foi mantido por sete dias. Foi feita cobertura, por esse prazo, com antibiótico, antinflamatório e tranqüilizante.
Como era de se esperar, também não conseguimos controlar o resultado pós-operatório pela rinomanornetria, apesar das muitas tentativas feitas. Todavia, após cinco meses de operado, uma inspeção feita sob sedação, com um endoscópio reto de fibras ópticas de Storz-Hopkins, revelou coarias permeáveis e de aspecto normal. Segundo sua mãe, depois da operação passara a dormir tranquilamente e sem ressonar.
COMENTÁRIOA imperfuração coaria( congênita é uma afecção relativamente rara e consiste no fechamento congênito da comunicação, normalmente existente, entre as fossas nasaisea rinofaringe. Atresia ou oclusão coaria( congênitas são designacoes sinônimas.
Extenso comentário sobre o assunto foi feito recentemente por um de nós (NEVES PINTO e DE BONA 1976). Assim, seria redundante voltarmos a discutir os vários aspectos desta afecção, não especificamente relacionados com o item agora tratado, isto é, a concomitância da imperfuração coana( com outras malformações congênitas.
Revisando a literatura sobre o tema em questão, McGOVERN (1950) encontrou 47 casos, relatados por 25 autores, nos quais as seguintes malformações congênitas, associadas à oclusão coana(, foram referidas: deformidade do palato duro, assimetria da face, úvula bífida, coloboma da íris, duplo tragus, fístula auricular, polidactilia e nariz bífido.
Outras associações congênitas, posteriormente relatadas, foram: imperfuração anal (SANTOS, AVELINO e MARQUES, 1972), malformações do aparelho gênito-urinário (BRIERRE JR., 1963), doença cardíaca (McGOVERN, 1953; CRAIG e SIMPSON, 1959; JOHNSEN; 1960), coloboma da íris e/ou retina (CRAIG e SIMPSON, 1959; FLAKE e FERGUSON, 1964), Síndrome de Treacher Collins (McNEIL e WINTER-WEDDERBURN, 1953; GRAHNE e KALTIOKALLIO, 1966), fístula cervical bilateral, assimetria da face, assimetria dos seios maxilares, malformação da asa nasal, malformação do crânio. hernia umbelical e inguinal, anus vestibular e microanus (GRAHNE e KALTIOKALLIO, 1966).
A esta lista, acrescentamos agora a associação entre imperfuração coanal congênita e Síndrome de Apert.
A Síndrome de Apert (APERT, 1906) é caracterizada pela presença de acrocefalia (produzida pela soldadura precoce das suturas) e sindactilia, às quais, mais das vezes, se soma retardamento mental. Outras anomalias associadas têm sido descritas, tais como: exoftalmia, exotropia, atrofia optica, oftalmoplegia, hipertelorismo, hipoplasia do maxilar superior com prognatismo relativo do maxilar inferior, palato ogival e fenda palatina.
As malformações cranianas, encontradas nessa -síndrome, são essencialmente as mesmas da Síndrome de Crouzon, a qual distinguimos da primeira pela ausência de malformações das mãos (v.g. GRABB e SMITH, 1970). Tratase de rara enfermidade congênita e de etiologia desconhecida. Um gene recessivo seria responsável pelos casos hereditários, mas entre 41 casos revisados por COMETTA e Cot. (1950) apenas 10 revelaram antecedentes familiares desta síndrome.
Também existe uma conotação entre hereditariedade e imperfuração coanal. Na literatura, são citados vários casos de oclusão coanal em gêmeos e parentes próximos (LANG, 1912; WRIGHT, 1922; PHELPS 1926; STEWART, 1931; SCHWECKENDIECK, 19(17; UMLAUF, 1939; BECKER, MATZKER e SCHIFFER, 1957; PARKES e BRENNAN, 1973). GRAHNE e KALTIOKALLIO (1966) acham que um gene defectivo pleiotrópico seria o responsável pelo aparecimento dessas malformações, em membros de uma mesma família.
O caso que vimos de descrever, além da acrocefalia, da sindactilia de todos os dedos de ambos pés e mãos, do retardamento mental e da imperfuração coanal unilateral, apresentava hipertelorismo, exoftalmia,- rinomegalia, deformidade septal, fenda palatina, hernia inguinal congênita, deformidade das fissuras e arcadas orbitárias superiores, soldadura precoce das cartilagens de conjugação dos membros superiores, malformações articulares dos ombros, cotovelos, pés e mãos, dentes mal implantados, hipoplasia de maxilar superior e prognatismo do maxilar inferior (várias dessas malformações foram corrigídas por cirurgia). Apesar de muitas das anomalias descritas serem meras consequências da malformação craniana, algumas são, fora de dúvida, independentes. Todavia, como GRAHNE e KALTIOKALLIO (1966), acreditamos numa causa primária comum a todas elas: um gene pleiotrópico defeituoso. 0 que não sabemos é se tal defeito (mutação) teve lugar durante a gestação do paciente ou se trata de característica hereditária de baixa dominância. Se não há antecedentes familiares de malformações congênitas, tampouco conseguimos identificar o uso de produto (s) químico (s) ou exposição a irradiação (por parte da mãe, durante a gestação) capazes de justificar uma mutação gênica.
SUMMARYCongenital Choanal Atresia and ApertYs Syndrome
The Authors report a case of complete ynilateral congenital choanal atresia in a patient vti'sth Apert's syndrome (Acrocephalo-Syndactyly). The malformation was corrected by surgery through the maxilla-premaxilla approach. They did not find any reference about this association in the consulted bibliography.
REFERÉNCIA BILBIOGRAFICASAPERT, E.: De L'acrocephalossyndactyllie. Bull. Soc. Med. Paris, 23: 1310, 1906.
BECKER, W., MATZKER, J. und SCHIFFER, K.H.: Ueber familiares Verkommen verschiedener Formen von Choanalatresie- Acta Otolaryng. (Stockh.), 47: 377, 1957.
BREIRRE JR., J.T.: Congenital abnormalities of the genRourinary tract: abdominal muscle displasia and choanal atresia. Pediatrics (EUA), 25: 290, 1963.
COMETTA, F., JUILLARD, E. et ROSSELET, P.: A propos d'un cas d'acrocephalosyndacttylie. Ophtalmologica (Brasl), 120: 72, 1950.
COTTLE, M.H., LORING, R.M., FISCHER, G.C. and GAYNON, LE.: The maxilla-premaxilla approach to extensive nasal septum surgery. Arch. Otolaryng., 68.
CRAIG, D.H. and SIMPSON, N.M.Posterior choanal atresia. J. Laryng., 73: 603, 1959.
FLAKE, C.G. and F?RGUSON, C-F.: Congenital choanal atresia in infants and children. Ann. Otol
Rhin. Laryng., 73: 458, 1964. GRABB, W.C. e SMITH, J.W.: Cirurgia plástica (otras anomalias da cabeza). Salvat Ed. S.A., Barcelona, 1970.
GRAHNE, B. and KALTIOKALLIO, K.: Congenital choanal atresia and its heredity. Acta Otolaryng. (Stockh), 62: 193, 1966.
JOHNSEN, S.: Congenital choanal atresia. Acta Otolaryng. (Stockh). 51: 533, 1960.
LANG, J.: Ueber choanenatresie Mschr. Ohrenheilk., 46: 970, 1912. McGOVERN, F.H.: The association of congenital choanal atresia and congenital heart d'ssease. Ann. Otol. Rhin. Laryng., 62: 894, 1953.
McGOVERN, F.H.: Congenital choanal atresia. Laryngoscope, 60: 815, 1950.
McNEIL, K. A. and WINTERWEDDERBURN, L.: Choanal atresia: a manifestation of the Treacher-Collins syndrome. J. Laryng., 67: 365, 1953.
NEVES PINTO, R.M.: Rinosseptoplastia funcional. Rev. Brasil. OtoRino-Laring., 41: 50, 1975.
NEVES PINTO; R.M. e DE BONA, C.: Imperfuração coanal congênita
unilateral: tratamento cirúrgico via maxila-Nremaxila. Rev. Brasil. Oto-Rino-Laring., 42: $$, 1976. PARKES, M.L. and BRENNAN, H.G.: Familial choanal atresia in a rhinoplasty candidate. Eye, ear, nose & thoroat, 52: 222, 1973. PHELPS, A.W.: Congenital oclusion of choanas. Ann. Otol. Rhin. Laryng. 35: 143, 1926.
SANTOS, M.A., AVELINO, L.C. de e MARQUES, V.P.B.: Imperfurações coanal (considerações
sobre dois casos). Rev. Brasil. Oto-Rino-Laring., 38: 215, 1972. SCHWECKENDIECK, H.: Transpalatine
Behandlung angeborener Choanalatresien. Z. Hals. Nas. Ohrenheilk., 42: 367, 1937. STEWART, J. P.: Congenital atresia of the posterior nares. Arch. Otolaryng., 13: 570, 1931. UMLAUF, I.: apud GRAHNE and KALTIOKALLIO (obra citada). WTIGHT, A.J.: Congenital occlusion of the posterior choanae. Lancet, 203: 569, 1922.
R.M. NEVES PINTO
Serviço Médico VARIG
Aeroporto Santos Dumont
Rio de Janeiro, R.J. 20.000
Brasil (tel.: 288-3470)
* Maj. Med. Aer. Assistente da CI. O.R.L. do H.C.Aer. (Rio de Janeiro). DocenteLivre de CIO.R.L. da F.M.P.A. (U.F.R.G.S.).
** 1º Ten. Med. Aer. Assistente da CI. O.R.L. do H.C.Aer.
