INTRODUÇÃODescrita por Waardenburgl, em 1951, esta Síndrome caracteriza-se pela associação de: deslocamento lateral do canto medial do olho (dystopia canthorum), heterocromia da íris, raiz nasal proeminente e alargada, hipertricose da porção medial dos supercílios, mecha branca frontal e surdez congênita neurossensorial uni ou bilateral.
Outras características têm sido descritas, relacionadas com a síndrome, como hipopigmentação cutânea, encanecimento precoce, hipoisocromia da íris, alterações de pigmentação retiniana2, associação com fendas palatinas e labiais e anomalias gastrointestinais4, como a doença de Hirschsprung e atresias do tubo digestivo5, 6.
A transmissão se dá por forma autossômica dominante, com penetrânciá e expressividade variáveis, sendo dividida em dois tipos7, 8 o tipo I, mais comum, com presença de dystopia canthorum, alterações faciais e perda auditiva bilateral; e o tipo II, menos comum, sem dystopia canthorum, sem alterações faciais, com mecha branca frontal comumente presente e envolvimento coclear mais freqüente que no tipo I9. Uma possível variante, autossômica recessiva já foi descrita4, 6, 10.
A característica mais comum é a dystopia canthorun; seguida pela mecha branca frontal. A raiz nasal proeminente geralmente segue a presença da dystopia (Tabela I). No entanto, o achado de maior importância, em razão das repercussões sociais, é a hipoacusia, na maioria das vezes bilateral e intensa e, em alguns pacientes, unilateral, raramente intensa11, 12. Estima-se que 2% a 5% dos pacientes com surdez congênita sejam acometidos por esta síndrome13, 14 incidência muito elevada para passar despercebida.
A descrição de um caso de criança surda, com albinismo parcial, defeitos em musculos esqueléticos e dystopia canthorum, por Klein, na mesma época que Waardenburg descreveu a síndrome, fez com que algumas vezes a SW seja denominada pela sinonímia Klein Waardenburg15, 18.
O objetivo do presente trabalho foi estudar as características de pacientes com diagnóstico de síndrome de Waardenburg, em acompanhamento no nosso serviço, com especial atenção ao problema auditivo.
MATERIAL E MÉTODOSForam avaliados oito pacientes com diagnóstico clínico de Síndrome de Waardenburg, em acompanhamento no Ambulatório de Otorrinolaringologia do Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto USP. Seis pacientes do sexo feminino e dois do sexo masculino; três leucodérmicos e quatro melanodérmicos; com idades variando de 5 a 60 anos (média de 21,12 anos). Quatro pacientes tinham classificação como SW tipo I, três SW tipo II e um sem classificação (Tabela II).
Todos foram submetidos a exame físico e otorrinolaringológico completo e audiológico, que constou de audiometria tonal liminar, imitanciometria e pesquisa do reflexo do estapédio contralateral. Os testes supraliminares e a audiometria de tronco cerebral não foram realizados devido à natureza das alterações auditivas encontradas (perda neurossensorial profunda bilateral ) e ausência de limiares para tal.
Exame eletronistagmográfico e pesquisa de anomalias do tubo digestivo não foram realizados devido à ausência de sintomatologia dos pacientes.
Para a realização da audiometria tonal, utilizou-se o audiômetro Pracitronic MA 31, em cabine acústica. Para a realização da imitanciometria e pesquisa do reflexo do estapédio, foi utilizado o aparelho Dicton CAT ZA42.
RESULTADOSA dystopia canthorum, característica mais comum e que define o tipo da Síndrome de Waardenburg, realizada pela medida do índice interocular, mostrou-se presente (índice > 0,6) em quatro pacientes (Síndrome de Waardenburg tipo I - SW I) e ausente (índice < 0,6) em três (Síndrome de Waardenburg tipo II - SW II). Um paciente perdeu o seguimento ambulatorial e não foi realizada nele a mensuração do índice inter-ocular (Tabela II).
A heterocromia de íris (Figura 1) foi encontrada em todos os pacientes com SW I (n=4) e em um paciente com SW II, perfazendo 62,5% do total. Dois pacientes com SM II apresentaram olhos azuis, sendo um deles melanodérmico.
A mecha branca frontal (Figura 2) foi encontrada em dois pacientes (25%), um deles com SW II e com presença também de albinismo parcial (Figura 3). O outro paciente mostrou-se com deficiência auditiva bilateral e sem medida do índice interocular.
A deficiência auditiva, encontrada em sete dos oito pacientes, foi do tipo neurossensorial profunda bilateral (tipo II de Hageman), com restos auditivos em algumas freqüências (graves ou agudas) nas intensidades de 85 a 115 dB, variando de paciente para paciente (Figura 4).
A presença de características clássicas ou isoladas da doença em parentes próximos ou distantes dos pacientes foi identificada em cinco dos oito indivíduos.
O resumo das principais características encontradas em cada paciente está abaixo:
Paciente 1 - V.C.S.
Sexo feminino, cor branca, 10 anos de idade, presença de dystopia canthorum, heterocromia de íris e deficiência auditiva profunda bilateral. Ausência de sinais em outros membros da família.
Paciente 2 - L.M.S.
Sexo feminino, cor branca, 17 anos de idade, presença de dystopia canthorum, heterocromia de iris e deficiência auditiva profunda bilateral. Ausência de sinais em outros membros da família.
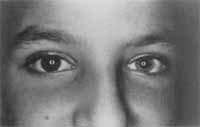
Figura 1. Paciente com heterocromia de íris.

Figura 2. Paciente com mecha branca frontal.
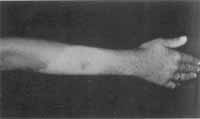
Figura 3. Paciente com albinismo parcial.
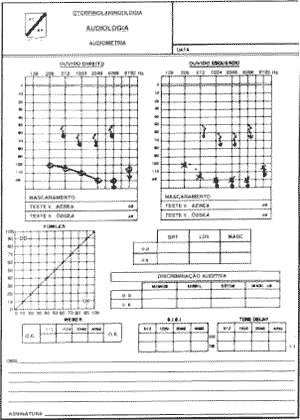
Figura 4. Audiometria tonal de um dos pacientes estudados.
Paciente 3 - R.S.B.
Sexo masculino, cor preta, 5 anos de idade, presença de dystopia canthorum, olhos azuis e deficiência auditiva profunda bilateral. A mãe apresenta heterocromia de iris, sem deficiência auditiva.
Paciente 4 - E.M.S.
Sexo feminino, cor preta, 60 anos de idade, presença de dystopia canthorum, heterocromia de íris, sem deficiência auditiva. Filho apresenta dystopia canthorum, olhos azuis e deficiência auditiva profunda bilateral.
Paciente 5 - E.L.B.
Sexo feminino, cor branca, 18 anos de idade, ausência de dystopia, presença de dois olhos azuis, albinismo parcial e mecha branca frontal. Ausência de sinais em outros membros da família.
Paciente 6 - M.J.R.S.
Sexo feminino, cor preta, 43 anos de idade, ausência de dystopia, presença de dois olhos azuis, deficiência auditiva profunda no ouvido direito e anacusia no ouvido esquerdo. História de bisavó com um olho claro e bisavô com dois olhos claros, sem deficiência auditiva.
Paciente 7 - A.V.T.
Sexo masculino, cor preta, 10 anos de idade, ausência de distopia, presença de heterocromia parcial de íris bilateral, deficiência auditiva profunda bilateral. História do pai apresentando mecha branca frontal e um tio com surdez não esclarecida.
Paciente 8 - T.G.O.R.
Sexo feminino, cor preta, seis anos de idade, dystopia não verificada, deficiência auditiva profunda bilateral e mecha branca frontal. Prima com mecha branca frontal e sem deficiência auditiva.
DISCUSSÃOA dystopia canthorum (aumento da distância entre os cantos internos das pálpebras, acompanhado do distanciamento dos pontos lacrimais inferiores, com distância interpupilar e entre os cantos internos dos olhos normal)16 é o achado mais consistente17, com penetrância estimada em 69%18. A presença de distopia pode ser comprovada pela medida do índice interocular a/b (a= distância entre os cantos internos; b= distância interpupilar), estabelecido por Partington19. O resultado acima de 0,6 comprova a existência da distopia18, 19. A presença de dystopia canthorum, encontrada em 4 pacientes (50%) foi a segunda característica mais freqüente da Síndrome de Waardenburg (SW), nos pacientes estudados.
A heterocromia da íris pode ser total, com íris de colorações diferentes, total ou parcialmente, quando parte de uma ou das duas íris apresentam colorações diferentes16. Alterações pigmentares retinianas têm sido descritas em correspondência com as variações de coloração da íris2, 13. Assim, íris hipocrômica está associada com fundo de olho hipopigmentado, íris mosqueada com fundo de olho mosqueado e íris castanha com fundo de olho castanho. A penetrância da heterocromia da íris é de 28%18. Existe ainda a hipoplasia da íris, que leva à hipoisocromia da mesma e que é considerada parte integrante da Síndrome2, 13, 18. A penetrância da hipoisocromia é de 29%18. A incidência de 50%, encontrada em nossos pacientes, difere dos 28% a 31% da literatura18, 20.
A mecha branca frontal pode ser discreta, constituída de apenas alguns fios ou mais acentuada. Pode ser evidente ao nascimento e tornar-se mais discreta com a idade ou preceder o encarecimento precoce. A penetrância é de 60%18. Esta característica foi encontrada em apenas dois pacientes (25%), incidência menor que a descrita na literatura18.
Áreas de pele com despigmentação em graus variados podem ser vistas em diversas partes do corpo, preferencialmente na região frontal, face anterior do tronco e extremidades dos membros13, 19. A penetrância é de 60%18. Áreas de despigmentação de pele localizadas, em troncos e membros, foram encontradas em apenas um paciente (12,5%).
A hiperplasia da porção medial dos supercílios e o alargamento da raiz nasal são alterações que comumente se associam16. A hipertricose de supercílios tem penetrância de 69%18. A raiz nasal alargada pode aparecerem 78% dos pacientes21.
O componente de maior importância clínica da Síndrome de Waardenburg é a hipoacusia. Os achados audiométricos, na síndrome de Waardenburg (SW), podem ser, divididos em dois grupos22:
- Grupo I: disacusia severa, com audição residual nas freqüências graves.
- Grupo II: disacusia moderada, com perda uniforme nas baixas e médias freqüências e audição melhor nas freqüências agudas.
As disacusias podem ainda ser classificadas em quatro tipos23:
- Tipo I: anacusia bilateral.
- Tipo II: disacusia severa bilateral.
- Tipo III: anacusia unilateral.
- Tipo IV: disacusia moderada unilateral, principalmente nas freqüências graves.
Waardenburg notou surdez neurossensorial congênita em 20% dos seus casos. Smith12 refere ser a surdez encontrada em 25% dos casos do Tipo I e 50% dos casos do Tipo II. Hangeman & Delleman20 encontraram 28% dos pacientes com síndrome de Waardenburg Tipo I (SWI) e 53% com síndrome de Waardenburg Tipo II (SWII), com surdez bilateral. Apenas 8% dos casos de SWI e 4% dos casos de SWII mostraram perda auditiva unilateral. O fato de sete dos oito pacientes apresentarem deficiência auditiva profunda bilateral (87,5%), incidência bem maior que os 36% encontrados por Hageman & Delleman20 na SW I, e os 57% na SW II, deveu-se ao fato de o nosso ambulatório ser referência para distúrbios da audição na região. Assim, os pacientes eram encaminhados devido ao distúrbio auditivo, na maioria das vezes sem diagnóstico feito e, a partir de então, era pesquisada a Síndrome, com identificação das outras características. Da mesma forma, o fato de todos os pacientes apresentarem deficiência auditiva profunda bilateral é conseqüência de, freqüentemente, a perda auditiva unilateral ou leve a moderada não serem detectadas.
A única paciente com SW em nosso estudo que não apresentava distúrbio auditivo foi identificada na anamnese de um dos indivíduos com a deficiência, por ser mãe do mesmo e apresentar outras características da síndrome, como heterocromia da íris.
Nestes casos, não foi possível detectar a progressividade ou não da perda auditiva. Porém, Hildesheimer & cols9 encontraram provável progressividade da perda auditiva em 70% dos pacientes estudados com SW II.
Jensen24 e Marcus25 descreveram estudos tomográficos do ouvido interno de pacientes surdos com SW, mostrando hipoplasia da cóclea, aplasia ou hipoplasia do canal semicircular posterior, vestíbulo anormal e ausência de janela oval. Nemansky & Hageman26, em estudo radiológico de 24 pacientes com SW (três com surdez bilateral, seis com surdez unilateral e 15 sem surdez), verificaram ausência de alterações anatômicas em todos eles. Fisch22 relata o exame post-mortem de uma garota com surdez profunda que faleceu aos três anos e meio de broncopneumonia, na qual havia ausência do órgão de Corti e atrofia do gânglio espiral e do nervo. No entanto, a causa da surdez parece estar realmente localizada no órgão de Corti, com atrofia do gânglio e do nervo da cóclea11, 12.
A possível explicação das diferentes manifestações da SW são algumas anormalidades que ocorrem no desenvolvimento da crista neural e das células que migram dessa estrutura27. O problema auditivo pode estar relacionado com a melanina. A principal estrutura em que a melanina é encontrada no ouvido interno é a ligth chromophobe cells da estria vascular, onde grânulos de melanina são freqüentemente vistos. Apesar de a função da melanina no ouvido interno ainda não estar estabelecida, alguns autores especulam sobre a importância de sua presença na elaboração, manutenção e reabsorção da endolinfa, o que poderia explicar a deficiência funcional no ouvido interno na SW9, 28.
A heterogeneidade genética da doença, com variável grau de expressividade e penetrância, faz com que os sinais característicos da Síndrome apareçam com freqüência e graus variados em cada indivíduo de uma mesma família afetada e nos acometidos em diferentes famílias.
As patologias vestibulares são encontradas na SW na mesma freqüência que em portadores de outros tipos de disacusias congênitas29.
A razão de três dos oito pacientes não apresentarem nenhuma característica da Síndrome em familiares pode ser atribuída a casos de mutação nova, estimada pela literatura em 25%3. Os demais pacientes parecem ter adquirido a. doença através da transmissão autossômica dominante.
O achado de incidência variável das características da Síndrome nos diferentes indivíduos estudados, ainda que da mesma família, deve-se à heterogeneidade da doença, com graus de expressividade e penetrância variáveis.
Para o aconselhamento genético, é importante ter em mente os levantamentos de Hageman & Deleman20, que, revendo 1.000, casos de SW descritos na literatura, concluíram que aproximadamente um quarto dos pacientes com SW I e metade dos pacientes com SW II têm perda auditiva neurossensorial bilateral congênita.
O aconselhamento genético é importante não apenas para prever o risco de transmissão da doença para novos indivíduos, a partir do paciente estudado, mas também para saber o que está acontecendo dentro da família do mesmo. Quando o paciente apresenta nova mutação, o risco para os pais é desprezível e o risco de transmitir a doença para um filho é de 50%. A probabilidade de a criança com a SW ter perda auditiva é de aproximadamente 25% para a SW I e de 56% para a SW II20.
Ainda que, em estudos das famílias individualmente, na literatura, a proporção das perdas auditivas bilaterais tenha sido maior que as perdas unilaterais, para aconselhamento genético, é melhor dizer que, se a perda auditiva ocorrer, é mais provável que seja bilateral do que unilateral30.
Um dos principais diagnósticos diferenciais deve ser feito com a síndrome de Woolf (piebaldismo associado à surdez). Outras duas síndromes que devem ser lembradas são a Síndrome de Vogt-Koyanagi (doença de Harada) e a Síndrome de Alezzandrini, ambas com alterações oculares e vitiligo, na primeira, e alterações auditivas, na última15.
CONCLUSÕESSíndrome de Waardenburg é doença que, com freqüência não muito rara, apresenta deficiência auditiva, na maioria das vezes profunda e bilateral.
O conhecimento das características da doença e sua identificação contribuirão para o diagnóstico etiológico precoce da perda auditiva relacionada com a mesma, evitando a realização de exames desnecessários, diminuindo os custos para o paciente ou a família do mesmo e favorecendo o início precoce da reabilitação auditiva.
O paciente portador da Síndrome de Waardenburg tem 50% de probabilidades de transmitir a doença para um filho. As probabilidades de ocorrência de problema auditivo na criança que desenvolveu a doença são de aproximadamente um quarto para a SW I e de metade para a SW II. Quando ocorre perda auditiva, é mais provável que seja bilateral do que unilateral.
AGRADECIMENTOSOs autores agradecemos, de forma especial, a Maria Rossato, pelo constante auxílio, dedicação e interesse durante a realização deste trabalho. E agradecem ainda ao Dr. Pércio Neves, Dr. Eduardo Scandiuzzi Pereira e Dr. Antônio Maria Claret Marra de Aquino, pela colaboração e apoio científico.
REFERÊNCIAS BIBLIOGRÁFICAS1. WAARDENBURG, P. J. - A new syndrome causining developmental anomalies of the eyelids, eyebrowns and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am. J Hum. Genet., 3: 195-253, 1951.
2. GOLDBERG, M. F. - Waardenburg's syndrome with fundus and other anormalies. Arch. Ophthal, 76.- 797-810, 1966.
3. PREUS, M.; LINSTROM, C.; POLOMENO, R. C.; MILOT, J. - Waardenburg syndrome - penetrance of major signs. Am. J. Hum. Genet., 15: 383-388, 1983.
4. FARNDON, P. A.; BIANCHI, A. - Waardenburg syndrome associated with total aganglionosis. Arch. Dis. Child., 58: 932-933, 1983.
5. NUTMAN, J.; STEINHERZ, R.; SIVAN, Y.; GOODMAN, R. M. - Possible Waardenburg syndrome with gastrointestinal anomalies. J Med. Genet., 175-178, 1985.
6. SHAH, K. N.; DALAL, S. J.; DESAIM, P.; SHETH, P. N.; JOSHI, N. C.; AMBANI, L. M. - White foreloch pigmentary disorder of irides and long segment Hirschsprung disease: possible variant of Waardenburg syndrome. J Pediatr., 99.432-435, 1981.
7. ARIAS, S. - Genetic heterogeneity in the Waardenburg Syndrome. Birth Defects: Original Article Series, 7: 87-101,1971.
8. HAGEMAN, M. J. - Audiometric findings in 34 patients with Waardenburg's Syndrome. J Laryngol. Otol., 9: 575-584, 1977.
9. HILDESHEIMER, M.; MAAYAN, Z.; MUCHNIK, C.; RUBINSTEIN, M.; GOODMAN, R. M. - Auditory and vestibular findings in Waardenburg's type II syndrome. J. Laryngol. Otol., 103: 1130-1133, 1989.
10. KULKARNI, M. L.; KURIAN, M.; GURUPRASAD, G.; PANCHAKSMARIAH, M. S. - Genetic heterogeneity in Waardenburg's Syndrome. J Med. Genet., 26: 411-412, 1989.
11. GORLIN, R. J.; PINDBORG, J. J.; COHEN, M. M. Syndromes of the head and neck. McGraw - Hill Book Company, New York, 1976.
12. SMITH, D. W. - Síndromes de malformações congênitas. Ed. Manole, São Paulo, 1985.
13. DI GEORGE, A. M.; OLMSTED, R. W.; HARLEY, R. D. Waardenburg's Syndrome: J Pediatr., 57: 649-669, 1960.
14. MARTINI, J.; CANT, J. S. - Waardenburg's Syndrome. Br. J Ophthalmol., 51: 755-759, 1967.
15. ROMITI, N.; DINATO, S. L. M.; GONZALES, M. L. S. Síndrome de Waardenburg. An. Bras. Dermatol., 62: 349-352, 1987.
16- ANTUNES, E. R. G.; ANTUNES NETO, E.; MATTAR, M. Síndrome de Waardenburg: estudo de uma família. An. Bras. Dermatol., 63: 75-84, 1988.
17. READ, A. P.; FOY, C.; NEWTON, V.; HARRIS, R. Localization of a gene for Waardenburg Syndrome type I. Ann. N Y. Acad. Sci., 143-151, 1991, 630p.
18. REED, W. B.; STONE, V. M.; BODER, E.; ZIPRKOWSKI, L. - Pigmentary disorders in association with congenital deafness. Arch. Dermatol., 95: 176-185, 1967.
19. PARTINGTON, M. W. - Waardenburg's Syndrome and heterochromia iridum in a deaf school population. Canad. Med. Ass. J., 90: 1008-1017, 1964.
20. HAGEMAN, M. J.; DALLEMAN, J. W. - Heterogeneity iri the Waardenburg's Syndrome. Am. J. Hum. Genet., 29: 468485, 1977.
21. MELO, L. R. P.; ALBERTINO, S.; TONON, S. - Syndrome de Waardenburg. Arq. Bras. Med., 58:147-150, 1984.
22. FISCH, L. - Deafness as part of an hereditary syndrome. J Hearing Otol., 73: 355-382,1959.
23. HAGEMAN, M. J.; GASTH, E. - Audiometric findings in 34 patients with Waardenburg's Syndrome. J. Laryngol. Otol., 91: 575-584, 1974.
24. JENSEN, J. - Tomography of the inner ear in a case of Waardenburg's Syndrome. Am. J. Roentgenol., 101: 828-833, 1967.
25. MARCUS, R. E. - Vestibular and additional findings in Wardenburg's Syndrome. Acta Oto-Laryngol (Suppl.), 229: 01-30, 1968.
26. NEMANSKY, J.; HAGEMAN, M. J. - Tomographic findings of the inner ear of 24 patients with Waardenburg's Syndrome. Am. J Roentgenol., 124: 250-255, 1975.
27. GOODMAN, R. M.; LEWITAL, I.; SOLOMON, A.; KLEIN, D. - Upper limb involvement in the Klein-Waardenburg Syndrome. Am. J. Med. Genet., 11: 425-433, 1982.
28. LA FERRIERE, K. A.; ARENBERG, I. K.; HAWKINS, J. E. Jr.; JOHNSSON, L. G. - Melanocytes of the vestibular labyrinth and their ralationship to the microvasculature. Ann. Otol. Rhinol. Laryngol., 83: 685-694, 1974.
29. HAGEMAN, M. J.; OOSTERNELD, W. J. - Vestibular findings in 25 patients with Waardenburg's syndrome. Arch. Otolaryngol., 103: 648-652, 1977.
30. NEWTON, V. - Hearing loss and Waardenburg's Syndrome: implications for genetic counselling. J. Laryngol. Otol., 104: 97-103. Feb., 1990.
* Médico Otorrinolaringologista - Pós-graduando de Otorrinolaringologia da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo.
** Professor Titular do Departamento de Oftalmologia e Otorrinolaringologia da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo.
*** Professora Assistente Doutora do Departamento de Oftalmologia e Otorrinolaringologia da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo.
**** Médica Otorrinolaringologista - Docente do Departamento de Fonoaudiologia da Faculdade de Filosofia e Ciências da UNESP - Marília.
***** Médica Assistente de Otorrinolaringologia do Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo.
Trabalho desenvolvido pelo Departamento de Oftalmologia e Otorrinolaringologia da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo.
Apresentado na XI Reunião da Sociedade Brasileira de Otologia e II Encontro Brasileiro de Trabalhos em Otorrinolaringologia, de 1° a 4 de novembro de 1995, em Belo Horizonte/ MG.
Endereço para correspondência: Dr. Thomaz J. M. Aquino - Rua Veiga Miranda, 188 /Apto. 21 - Bairro Jardim Mosteiro - CEP: 14085-200 - Ribeirão Preto/ SP.
Artigo recebido em 12 de agosto de 1996. Artigo aceito em 30 de outubro de 1996.


