INTRODUÇÃOEm 1914, Usher descreveu uma síndrome de natureza genética que se caracterizava, sobretudo, por 2 grandes sintomas: surdez congênita bilateral e cegueira progressiva, que levava seu portador a abandonar suas atividades profissionais a partir dos 40 anos, devido a grande perda da capacidade visual.
A retinite pigmentar é bastante freqüente, segundo Barkur (1994), atingindo um milhão e meio de pessoas; mas, associada a hipoacusia, é patologia rara na população (3 por 100.000 habitantes) [Fraser (1964) e Hallgrenn (1959)]. Dentro do sub-grupo dos portadores de surdez congênita, sua incidência é bastante expressiva (3 a 6%), segundo Fraser (1964) e Liebreich e col. (1964). Estudo realizado por Parnicky (1957) em crianças surdas e cegas nos Estados Unidos revelou que 2/3 das mesmas apresentavam a Síndrome de Usher. Segundo Hallgrenn (1959), esta síndrome tem incidência maior nos indivíduos da raça judaica e naqueles conseqüentes dos casamentos consangüíneos.
Segundo a maioria dos autores, a síndrome de Usher produz cegueira progressiva por retinite pigmentar, surdez congênita, perda do olfato e do paladar, lesões no sistema vestibular, alterações em todo o sistema nervoso central, distúrbios psiquiátricos como alucinações, psicoses variadas, alterações paranóicas, retardo mental etc.
Acredita-se que esta síndrome seja transmitida por gen autossômico recessivo, sendo por isso que, em 100 pessoas portadoras, somente 3 apresentaram os sintomas da doença. Entretanto, Scott & Donnell (1995) acreditam ser ela causada por falha autossômica dominante, o que explicaria sua incidência em vários membros da mesma família. Mas, em realidade, existe ainda muita controvérsia, se ela seria mesmo doença genética única ou se a mesma seria devida à conjugação de várias condições (McGovern, 1960). Isto explicaria a possibilidade de termos a síndrome de Usher com retinite pigmentar com e sem surdez, e talvez sem a retinite. McCay, em 1969, descreveu que em 8 casos, havia dois pares de irmãos; portanto, 50% de sua casuística era tipicamente hereditária. Segundo Bossu & Luypaert (1958), ela é devida à degeneração da retina e parte do cérebro pelo acúmulo de certos lipídios nas células do sistema nervos o que justificaria a perda de visão, de audição, do olfato, do paladar e as alterações neurológicas e psiquiátricas.
Existem inúmeros trabalhos sobre esta moléstia literatura oftalmológica, neurológica, psiquiátrica, genética etc, mas ela é ainda muito pobre quando se refere à otorrinolaringológica. Tanto isto é verdade, que alguns autores descrevem esta síndrome como surdez congênita, enquanto outros, como McCay, (1969) acreditam que ela seja progressiva. Alguns acham que a evolução da moléstia leva à perda total da audição (Parnicky, 1959), enquanto outros como (Froggatt 1964), acreditam que sempre irá permanecer algum resíduos auditivo.
As alterações no sistema vestibular são também bem conflitantes. O acometimento das vias vestibulares varia de 0 a 100%, mas parece que 90% sejam o valor mais correto (Hallgrenn 1959, Landau & Freitunesser, 1956 e Cremonesi e Mora, 1962). Quanto ao local lesado, também existe controvérsias sobre patologia labiríntica e/ou cerebelar.
A perda do olfato e, em alguns casos, do paladar (Bossu Luypaert, 1958), ainda permanece obscura devido à dificuldade à se avaliar, ainda hoje, quantitativamente estas funções. Entretanto este autores acreditam que essas alterações sejam de natureza central.
APRESENTAÇÃO DO CASOPaciente RL, masculino, 54 anos, casado, engenheiro. Encaminhado pelo Serviço de Oftalmologia por apresentar grande perda da acuidade visual progressiva (retinite pigmetar), zumbidos e surdez há algum tempo.
O paciente vem há mais de 6 meses apresentando zumbidos fortes em ambos os ouvidos, com nítido predomínio do lado direito com perda auditiva bilateral neste período. Nega tonturas. Nega alterações do olfato e do paladar. Nega dores na nuca. Pressão arterial levemente aumentada há anos, mas controlada com medicamentos (betabloqueador). Antecedentes pessoais: nada digno de nota. Tem irmã com quadro semelhante, porém nela a perda auditiva é bem mais discreta, mas em contrapartida a visual é bem mais intensa. Tem outro irmão sem qualquer alteração. Os pais não tinham qualquer parentesco entre si e não eram de descendência judaica.
Paciente com bom relacionamento, não demonstrando possuir alterações significativas no aspecto psiquiátrico. Apresenta distúrbio de fala característica dos hipoacúsico: antigos.
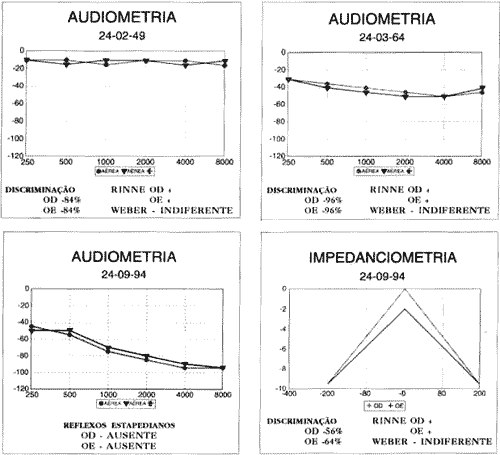
Exame ORL normal. Audiometria em 24/09/94, por ocasião da consulta, demonstrou perda auditiva bilateral de grande intensidade, com queda nos agudos. Imitanciometria normal, sem reflexos estapedianos. Em sua ficha antiga, constavam duas consultas anteriores com dois testes audiométricos: a primeira em 08/02/49, quando possuía 9 anos de idade, apresentou audiometria normal e a segunda consulta, em 24/03/64, aos 24 anos, mostrou discreta hipoacusia bilateral.
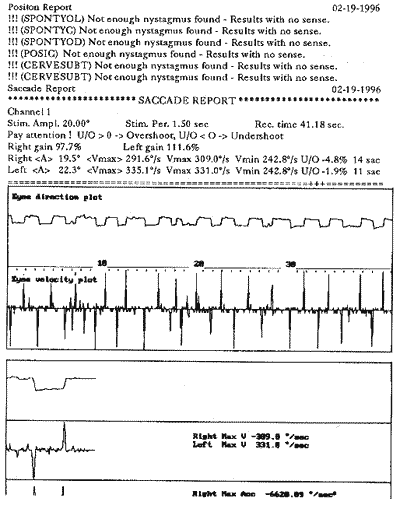
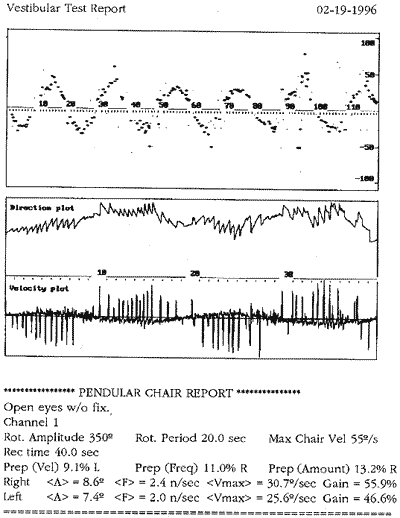
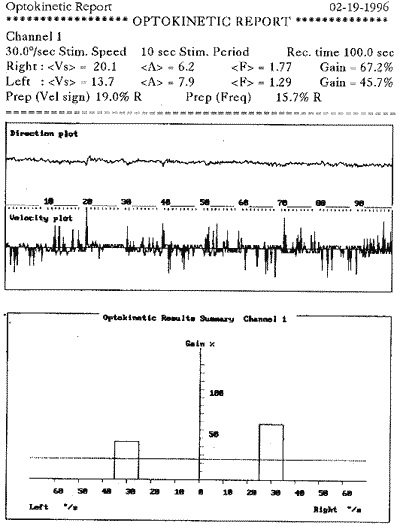
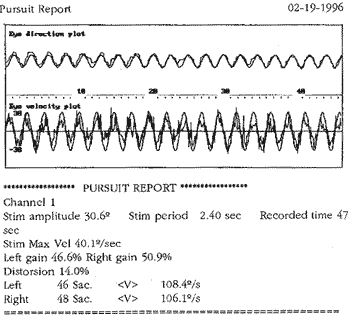
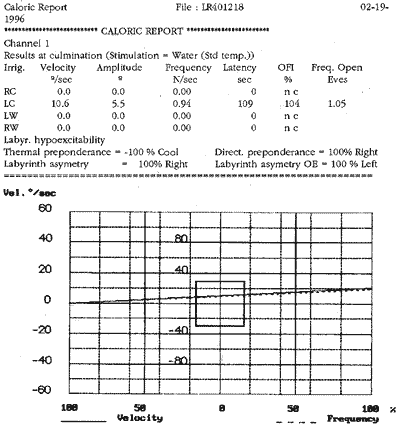
Ao exame otoneurológico, não se constataram sinais espontâneos, os testes de oculomotricidade foram aparentemente normais (prejudicados pela diminuição da acuidade visual), a prova pendular rotatória estava dentro dos padrões da normalidade, o índice de fixação não foi realizado pelo mesmo problema visual e, finalmente, as provas calóricas revelaram quase ausência de respostas. Concluiu-se pela síndrome deficitária bilateral periférica.
COMENTÁRIOSAnalisando os dados obtidos neste paciente, confirma-se o diagnóstico de Síndrome de Usher: caráter genético, perda visual por retinite pigmentar e hipoacusia bilateral. Entretanto, sua hipoacusia não foi congênita, mas provavelmente progressiva e com agravamento na quinta década, o que concorda com os dados obtidos por McCay (1969). O sistema vestibular estava comprometido por lesão tipicamente periférica, contrariando os dados de Cremonesi Mora (1962). Não foram encontradas as alterações sensoriais (olfato e paladar), neurológicas e psiquiátricas descritas por diversos autores.
Supondo-se que seu irmão também apresentasse a síndrome de Usher, pode-se admitir que esta moléstia não seria uma única doença genética, mas sim conjugação de condições variadas que levaria à diversificação dos sintomas e também ao modo de aparecimento dos mesmos; concordando com os trabalhos de McGovern (1960).
APRESENTAÇÃO DOS EXAMESREFERÊNCIAS BIBLIOGRÁFICAS1. USHER, C. - On the inheritance of retinitis pigmentosa, with notes of cases. Royal London Ophth. Rept. 9, 130, 1914 in MacCay Usher's Syndrome. J. Chron. Dis. 22, 133, 1969
2. BARKUR, S. - Retinites pigmentosa and related disorders. Am. J. Med. Genet. 52, 467, 1994.
3. FRASER, G. - Profound childhood deafness. /. Med. Genet. 1, 118, 1964.
4. HALLGRENN, B. - Retinitis pigmentosa combined with congenital deafness. Acta Psychiat. Scand. 34, 138, 1959.
5. LIEBREICH, R. - Abkuntf aus Ehen Unter Blutsverwandten als Grand von Retinites Pigmentosa. Dt. Klin. 13, 1961 in Fraser, G. - profound chilhood deaness. /. Med. Genet. 1, 118, 1964.
6. PARNICKY, J. - Characteristics of deaf-blind adults at the idustrial honre for the blind in McCay, v. - Usher's Syndrome. J. of Chronic Diseases, 22, 133, 1969.
7. SCOTT, R. & DONNELL, C. - Pattern dystrophy and retinites pigmentosa caused by a peripherin/RDS mutation. Retina, 15, 68, 1995.
8. Mac-GOVERN, F. - Association of nerve deafness and retinites pigmentosa. Ann. Otol. Rhin. Lar. 69, 1044, 1960.
9. McCay, V. - Usher's syndrome - deafness and progressiva blidness. J. Chronic Diseases 22, 133, 1969.
10. Bossu, A. & Luypaert, R. - The syndrome od Usher. Annal. Oculist., 191, 529, 1958.
11. Frogatt, P. - Public health and congenital disease: The example of congenital deafness. Irish J. Med. Soc 57, 1964 in McCay, V. - Usher's Syndrome. J. Of Chronic Diseases, 22, 133, 1969.
12. Landau, J. & Feinmesser, M. - Audiometric and vestibular examinations in retinitis pigmentosa. Br. J. Ophtal. 40, 40, 1956.
13. Cremonesi, G. & Mora, E. - The Usher's syndrome. Arch. Ital. Lar. 70, 427, 1962.
* Professores Doutores.da F. C. M. Unicamp - Campinas.
** Acadêmica de Medicina da Pucamp - Campinas.
Trabalho realizado na Clínica de ORL do Instituto Penido Burnier - Campinas.
Endereço para correspondência: O. Maudonnet Av. Andrade Neves, 611 - Campinas - SP CEP 13013-161; Fone: (019) 236.1027.
Artigo recebido em 29 de fevereiro de 1996. Artigo aceito em 15 de abril de 1996.