INTRODUÇÃOPoucas vezes na recente história da medicina se avançou tanto, em tão pouco tempo, como no campo da biologia molecular. Como bem define Post, biologia molecular é o estudo do fluxo de informações através da célula. Essas informações controlam a futura diferenciação e atividade celular. O DNA é o local de armazenamento de toda essa informação, que posteriormente é transcrita no RNA mensageiro. A informação então flui do núcleo para o citoplasma, alcançando os ribossomos, onde é traduzida em proteínas, as quais executam o trabalho da célula.
A genética molecular é uma subdivisão da biologia molecular e se ocupa do estudo da hereditariedade na transmissão da referida informação, sempre que esta apresenta algum defeito, que se traduz em uma doença ou fenótipo alterado. A pesquisa da localização cromossômica de genes, do isolamento de genes propriamente ditos, da detecção de mutações genéticas e das conseqüências dessas mutações, tanto em termos de funcionamento celular quanto de determinação de fenótipos, faz parte do campo da genética molecular.
Esse artigo objetiva revisar os recentes avanços na área da genética molecular da surdez. É cada vez mais importante que o otorrinolaringologista domine esse campo de conhecimento, tanto para poder entender a moderna fisiologia coclear quanto para melhor aconselhar, diagnosticar e, num futuro próximo, tratara surdez hereditária.
EPIDEMIOLOGIA E DEFINIÇÕES DA SURDEZ HEREDITÁRIA
Aproximadamente uma em cada 1.000 crianças é afetada por surdez severa ao nascer ou até o término do período pré~ lingual. Mais uma em cada 1.000 crianças torna-se surda antes de alcançar idade adulta. Finalmente, 0,3% da população manifesta perda auditiva acima de 65 dB entre 30 e 50 anos; e 2,3%, entre 60 e 70 anos. A prevalência continua a aumentar e alcança índices de 50% em octagenários2.
Apesar da grande importância da perda auditiva em idade avançada, na maioria os trabalhos realizados procuraram estudar formas de surdez pré-lingual, quer pela maior severidade do quadro, quer pela maior facilidade diagnostica. Contudo, mesmo com adição do efeito de fatores ambientais, não se pode excluir susceptibilidade genética nas perdas de aparecimento tardio.
Atualmente, 60% dos casos de surdez pré-lingual em países desenvolvidos são devidos a defeitos genéticos j. Em países em desenvolvimento, como o Brasil, causas infecciosas ainda são bastante comuns, mas esse número tende a diminuir com a melhoria gradativa das condições de saúde pública. Assim, a etiologia genética tende a se tornar cada vez mais importante também entre nossos casos de surdez.
A surdez hereditária pode ser classificada em sindrômica e não sindrômica (isolada). As formas ditas sindrômicas perfazem aproximadamente 30% dos casos de surdez hereditária em crianças4, e o déficit é na grande maioria condutivo ou misto (condutivo + sensorioneural). Algumas centenas de síndromes foram descritas onde surdez é uma das anomalias associadas. Grande proporção destas consiste em defeitos na formação embriológica do ouvido, e aproximadamente 60 genes implicados nestas síndromes já foram mapeados no genoma humano. Mais da metade destes já foram clonados. O Quadro 1 traz as síndromes mais conhecidas com seu mapeamento e o gene mutado responsável (Quadro 1). Uma contribuição de pesquisadores brasileiros aparece no estudo de ligação, publicado em 1998, realizado em família com síndrome de Bjornstad (associação de surdez sensorioneural congênita e malformação peculiar do cabelo - pili torti). Mapeou-se o gene responsável no locus 2834-36 (braço longo do cromossomo 2, entre as bandas 34e36)5.
Em contraste, a busca por genes responsáveis pelas formas não sindrômicas de surdez está apenas começando. Esta revisão se concentra nestas últimas, uma vez que são estas as mais prevalentes na população de crianças com surdez hereditária (70% dos casos) e na clínica diária.
MODELOS DE MAPEAMENTO GÊNICOTrês diferentes modelos vêm sendo utilizados para se mapear os genes responsáveis pelas formas genéticas de surdez não sindrômica. O primeiro consiste no estudo de grandes famílias afetadas onde exista alta taxa de consangüinidade. Geralmente, estas famílias são encontradas em regiões geograficamente isoladas, onde têm permanecido por várias gerações. Neste modelo, a probabilidade de que mais de um gene possa estar causando doença é minimizada, reduzindo a assim chance de heterogeneidade genética. O estudo é realizado através do mapeamento genômico dos indivíduos destas famílias em busca de regiões do DNA compartilhadas apenas pelos indivíduos afetados. Este foi o modelo utilizado por nós para o mapeamento da síndrome de Bjornstad. Estudando-se uma família mexicana com história de consangüinidade com oito indivíduos afetados, delimitou-se uma região de 3 cm na telomérica do braço longo do cromossomo 2, onde não havia recombinações genéticas entre os afetados5.
A segunda abordagem se clã através do estudo de populações isoladas de fluxos migratórios, onde estudos de segregação apóiem um modelo autossômico dominante. O estudo é então realizado através da busca de alelos nos indivíduos afetados que estejam em desequilíbrio com relação à freqüência destes na população em geral.
Finalmente, diversos genes responsáveis por formas não sindrômicas de surdez já foram localizados através do estudo de pequenas famílias com indivíduos afetados e a busca de marcadores que sejam homozigóticos nestes. Contudo, este último modelo pressupõe ou uso de mapas genômicos muito densos (o que pode tornar impraticável qualquer tentativa de identificação), ou que o gene candidato para doença seja previamente definido.
QUADRO 1 - Principais síndromes que envolvem perda auditiva já descritas molecularmente.
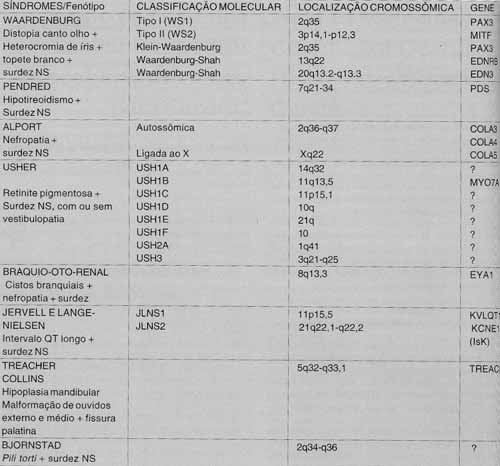
Legenda: p- braço curto do cromossomo; q- braço longo do cromossomo; 2q- braço longo do cromossomo 2; 2p34-36- braço longo do cromossomo 2 entre as bandas 34 e 36.
Para formas autossômicas dominantes de surdez não sindrômica, vários genes já foram identificados através do estudo de grandes famílias vivendo em regiões geograficamente isoladas. Deve se deixar claro, contudo, que não existe necessidade a priori de que estas famílias vivam em regiões isoladas. Este fato, além de aumentar a chance de que formas recessivas da doença apareçam, diminui a chance de que uma mesma doença possa estar sendo causada por dois diferentes genes (heterogeneidade genética).
QUADRO 2- Formas de surdez genética não-sindrômica de transmissão autossômica dominante.
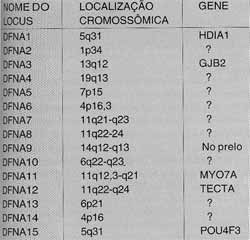
Legenda: DFNA - Formas de perda auditiva não sindrômica de transmissão autossômica dominante.
Além dos métodos já existentes de clonagem posicional, a identificação destes. genes pode ser facilitada através do achado de alterações cromossômicas (como deleções ou translocações), como foi o caso, por exemplo, da síndrome de Waardenburg. Outra forma que contribui para identificação de genes é o estudo de modelos animais já existentes para surdez. Vale lembrar que o DNA tem estrutura bastante conservadora entre espécies e que é possível estabelecer com alto grau de certeza homologia entre DNA do camundongo e o humano. Atualmente, mais de 25 tipos de camundongos geneticamente modificados são modelos animais para diferentes tipos de perda auditiva, inclusive sindrômica 6,7.
QUADRO 3 - Formas de surdez genética não-sindrômica de transmissão autossômica recessiva.

Legenda: DFNB -Formas de perda auditiva não sindrômica de transmissão autossômica recessiva.
Cerca de 85% dos casos de surdez pré-lingual não sindrômica manifestam-se como formas autossômicas recessivas. Formas autossômicas dominantes respondem por 12 a 14% dos casos e os restantes 1 a 3% são heranças mendelianas ligadas ao cromossomo X 8,9. Também se descrevem formas herdadas exclusivamente através da mãe, correspondendo à herança mitocondrial, associadas ou não à herança autossômica dominante.
Em termos fenotípicos, formas autossômicas recessivas são mais severas, sendo responsáveis por aproximadamente todas, as formas de surdez congênita. Na maioria das vezes, são devidas a defeitos cocleares. Formas autossômicas dominantes parecem contribuir mais importantemente para casos de surdez pós-lingual. Essas últimas são geralmente progressivas, menos severas (pelo menos nos anos iniciais de aparecimento) e podem mostrar associação de déficits tanto condutivos quanto sensorioneurais 8,9.
Arbitrariamente, convencionou-se chamar diferentes localizações cromossômicas (locus, do latim; plural, loci) das formas não sindrômicas de surdez genética com a sigla DFN (oriunda do inglês deafness) acrescida ou não das letras A e B, significando forma de transmissão autossômica dominante (DFNA) e recessiva (DFNB), respectivamente. Quando aparecer DFN isoladamente, leia-se surdez de transmissão ligada ao cromossomo X. Até o momento, já foram descritas 15 formas de perda auditiva não sindrômica de transmissão autossômica dominante (DFNA);19, de transmissão autossômica recessiva (DFNB); 6, de transmissão ligada ao cromossomo X (DFN); e 2 de transmissão mitocondrial. Os Quadros 2, 3, 4 e 5 retirados com algumas modificações da Hereditctry Hearing Loss Homepage 11, descrevem forma genética de transmissão, loci e genes, para diferentes formas de surdez não sindrômica.
QUADR0 4 - Formas de surdez genética não sindrõmica de transmissão ligada ao cromossomo

Legenda: DFN - Surdez de transmissão ao cromossomo X.
QUADRO 5 - Formas de surdez genética não sindrõmica de transmissão mitocondrial.

Uma breve revisão da fisiologia coclear normal se impõe para se entender as conseqüências das mutações dos genes já mapeados no processo de audição. Após estímulo sonoro, energia mecânica do som é convertida em sinal elétrico (transdução mecanoelétrica) nas células ciliadas do órgão de Corti.
Na superfície apical dessas células ciliadas, projetam-se microvilosidades especializadas, estereocílios, em arranjos organizados em forma de V. Estruturalmente, estereocílios consistem de cório com activa e revestimento externo de miosina. Eles oscilam em reposta ao som, que, secundariamente ao pistoneamento do estribo na janela oval, movimenta o líquido que circunda as células ciliadas.
É essa deflexão dos estereocílios adjacentes que abre os canais de transdução, permitindo um influxo de potássio da endolinfa para células ciliadas, causando despolarização da membrana celular. A despolarização ativa canais de cálcio da superfície basolateral da membrana que são sensíveis às alterações de voltagem, e subseqüente influxo de cálcio provoca liberação de vesículas contendo neurotransmissores nos terminais sinópticos dó VIII nervo craniano.
Dessa forma, após estimulação sonora, células ciliadas ficam hiperpolarizadas, com alta concentração de potássio intracelular. Para que nova excitação da célula seja possível, potássio tem que ser removido. É muito provável que tal movimento de íons potássio das células ciliadas de volta para endolinfa se faça através de comunicações intercelulares especializadas entre células ciliadas e células de sustentação do órgão de Corti, junções do tipo gap. Essas junções são cilindros constituídos por proteínas especiais, as conexinas. Em última análise, é esse movimento do potássio o responsável pelo potencial endococlear13.
Então, na cóclea, mutações de genes que codificam proteínas componentes do aparato de transdução, que afetam estrutura do estereocílio e estejam envolvidas na liberação da vesícula sinóptica ou participem no transporte de potássio, podem potencialmente resultar em perda auditiva.
GENES RECENTEMENTE MAPEÀDOS E SEQÜENCIADOS DE PERDA AUDITIVA NÃO SINDRÔMICAEstimou-se o número total de genes responsáveis por perdas auditivas não sindrômicas entre 30 e 100 genes, mas devemos levar em consideração o pouco conhecimento molecular dos processos de transcrição gênica e sinalização envolvidos na audição.
0 total de oito genes responsáveis por surdez já foram identificados, seqüenciados e descritos. Outros genes já identificados estão em vias de publicação. Por exemplo, participamos do seqüenciamento e da identificação de três diferentes mutações no gene para a DFNA9, uma forma de surdez de aparecimento tardio que progride para cofose, em que membros afetados de três diferentes famílias demonstram depósitos eosinofílicos (semelhantes a mucopolissacarídeos) no estudo histopatológico do ouvido interno12. (O artigo do seqüenciamento do gene para DFN9 está em vias de publicação.)
Mutações em genes de dois tipos de miosinas nãoconvencionais, miosina 15 (MYO15) e miosina VILA (MY07A), foram identificadas como responsáveis por surdez genética. As miosinasnão convencionais são "motores moleculares" que se ligam à activa, hidrolizam ATP e deslocam-se ao longo dos filamentos de activa. A miosina 15 está ïmplicada em forma recessiva não sindrômica de perda auditiva, DFNB3 13,14 A miosina VIIA é reponsável por forma autossômica recessiva (DFNB2)15, e outra forma autossômica dominante (DFNA11)16, além de ser o gene mutado na síndrome de Usher (tipo 1b)17. No ouvido interno, este último tipo de miosina está presente tanto nas células ciliadas internas quanto nas células ciliadas externas e sua função parece estar relacionada com estrutura dos estereocílios e com transdução de sinal durante processo de audição. O arranjo em forma de V dos estereocílios é importante para permitir abertura sincronizada dos canais iônicos durante vibração induzida pelo movimento de endolinfa. Mas em ratos com mutações na miosina VIIA, ao invés da conformação típica, o que se vê são estereocílios organizados anarquicamente, em pequenos tufos 18. A miosina VIIA pode carrear ou ancorar outras moléculas que são importantes para formar o arranjo - preciso dos estereocílios, além de já ter sido associada com liberação de vesículas sinápticas 19.
A Conexina 26 (também chamada GJB2), um dos genes que codifica proteína da família das conexinas, já foi identificada como contendo mutações em formas autossômicas dominantes (DFNA3) e recessivas (DFNB1) de perda auditiva20,21. As junções do tipo gapestão envolvidas no processo de criação e manutenção do potencial endococlear. Um defeito na estrutura e funcionamento dessas comunicações intercelulares poderia causar uma manutenção de altas concentrações de potássio intracelular, o que prejudicaria o mecanismo que permite resposta rápida da célula ciliada ao novo estímulo sonoro20. A conexina 26 parece ser gene extremamente comum na gênese da surdez hereditária. Em trabalho recente, 65 famílias com história de surdez provenientes de vários países (Tunísia, França, Nova Zelândia e Reino Unido) foram testadas para mutações na conexina 26. Em 39 dessas encontraram-se mutações nesse gene. O que chamou mais atenção é que, em metade delas, a mutação foi a mesma 22. Esse achado, associado ao fato de a conexina 26 ser um gene com estrutura extremamente simples (contendo somente uma região codificadora), justifica previsão otimista de que será relativamente simples de se desenvolver teste diagnóstico genético de uso clínico corriqueiro para detectar mutações na conexina 26. Tal teste poderá permitir diagnóstico rápido de surdez em recémnascidos, permitindo, no mínimo, intervenção precoce. O HDIA1 (também chamado de diaphanous), é gene da família das forminas responsáveis pela definição dá polaridade celular. Foi identificado como responsável pela primeira forma de surdez não sindrômica descrita na literatura (DFNA1)23. Parece que a proteína codificada por esse gene serve como um suporte temporário para actina, quando ela se rearranja para ajudar a célula a se dividir ou formar projeções, como os estereocílios.
Outro gene já identificado como causador de perda auditiva não sindrômica, POU3F4, codifica uma proteína relacionada com transcrição de DNA (fator de transcrição), que desempenha importante papel na regulação do desenvolvimento de tipos celulares. Mutações neste gene foram encontradas em forma ligada ao cromossomo X (DFN3)24 . Gene com função semelhante, POU4F3, quando mutado, é responsável pela forma autossômica dominante DFNA1525.
O gene TECTA, assim chamado por ser responsável pela síntese de proteína estrutural da membrana tectorial do orgão de Corti, alfa-tectorina, demonstrou mutações em duas formas de surdez genética, DFNA8 e DFNA12 26. A membrana tectórica é uma matrix extracelular do ouvido interno que
contata com feixes de estereocílios de células ciliadas. A alfatectorina é um dos maiores componentes não colágenos da membrana tectõrica.
Novas mutações no gene PDS, assim denominado por tratar-se de gene mutado na síndrome de Pendred27, foram encontradas nos afetados por forma de surdez autossômica recessiva, DFNB428.
O FGFR3, gene que codifica um dos subtipos de receptores para fator de crescimento de fibroblastos, no qual mutações haviam previamente se mostradas associadas à craniosinostose e a outras malformações esqueléticas, foi recentemente proposto também como gene candidato para perda auditiva sensorineural do tipo autossômico dominante (DFNA6)29. Mutações em famílias ligadas a este intervalo, contudo, ainda não foram descritas.
Ainda, duas mutações em genes do DNA mitocondrial, genes 12S rRNA e tRNA(ser) UCN também predispõem à perda auditiva relacionada com idade. Em particular, mutação A1555G no gene 12S rRNA foi relacionada com maior susceptibilidade à perda auditiva pós uso de antibióticos aininoglicosídeos e pode ser importante causa de perda progressiva da audição em algumas populações, mesmo na ausência da exposição a aminoglicosídeos 3°.
Resumindo, na cóclea, mutações de genes que codificam proteínas componentes do aparato de transclução, que afetam a estrutura do estereocílio, que estejam envolvidas na liberação da vesícula sinóptica ou que participem no transporte de potássio podem resultar em perda auditiva.
A clonagem dos genes responsáveis por todas as diferentes formas de perda auditiva, embora caminhe a passos largos, continua sendo um grande desafio. Devido a grande heterogeneidade genética e clínica na apresentação cia doença, a definição de intervalos genômicos associados com doença é mais facilmente realizada, com estudo de grandes famílias onde a surdez é segregada através de diferentes gerações. Uma vez identificados, estes intervalos ainda contêm um grande número de genes, ou mesmo de transcritos ainda não identificados. Isto explica porque, à exceção dos poucos casos mencionados, todos genes responsáveis por estas condições ainda não foram seqüenciados.
IMPLICAÇÕES CLÍNICAS E ÉTICASAtualmente, como nem todas as mutações que podem causar surdez estão identificadas, ainda é difícil rastrear-se sistematicamente mutações em pessoas cota história familiar de perda auditiva, a menos nos poucos casos onde o gene envolvido na afecção já tenha sido determinado. Ou seja, a exclusão de determinada mutação potencialmente relacionada com sudez não diminui muito o risco de determinado indivíduo com história familiar desenvolver perda auditiva no futuro, uma vez que outros defeitos podem estar implicados na gênese de perda auditiva da família ou do indivíduo analisado.
Já existem tentativas no sentido de se poder prever com mais confidência se determinado indivíduo, no contexto de sua família, herdou ou não defeito genético causador de surdez. Chen e colaboradores, em recente trabalho, estudaram a possibilidade de se prover diagnóstico pré-sintomático para indivíduos jovens pertencentes a famílias participantes do estudo onde a localização genômica do defeito causador da afecção (no caso, perda auditiva não sindrômica do tipo autossômica dominante) havia sido previamente determinada. Os resultados foram animadores, e o uso das técnicas de análise do DNA permitiu prever com maior acerto a probabilidade de surdez nos afetados31.
Alguns autores advogam teste genético em indivíduos com perda auditiva induzida por aminoglicosídeos para presença da mutação mitocondrial AI 555G, uma vez que sua presença permitiria aconselhamento de todos os descendentes maternos. Sendo teste facilmente realizável e o aconselhamento possível, esta prática poderia ser inclusive custo-efetiva 32.
O diagnóstico pré-natal também passa a ser possível, embora ainda seja motivo de controvérsia 33. Sérias dúvidas sobre a real validade desta aplicação se encontram tanto na ética médica quanto nos valores morais da sociedade. O ponto central da discussão é a possibilidade ou não de término da gestação quando de um resultado não favorável nos testes de rastreamento genético. Da mesma forma, o resultado "não favorável" pode ser entendido no sentido inverso. É o que ocorre, por exemplo, nos Estados Unidos, onde alguns membros da comunidade surda respondem que preferem filhos surdos do que ouvintes. A lógica adotada é a de que a surdez não é uma doença que requer tratamento; mas, sim, mais um dos aspectos de cultura diferente. A comunidade surda tem linguagem única (sinais), assim como crenças, costumes e valores próprios. Membros dessa comunidade podem ter objetivos a serem alcançados, diferentes dos do aconselhamento genético, e não necessariamente vêem a surdez com condição que requeira tratamento.
Com identificação de novos genes responsáveis por perda auditiva a cada mês, testes diagnósticos pré-sintomáticos serão cada vez mais parte do dia-a-dia do otorrinolaringologista, de seus pacientes e das famílias destes. Cabe a cada um preparar-se para que os avanços da genética médica sejam utilizados da melhor forma possível e não como instrumentos de discriminação ou abuso, como, por exemplo, a negação de cobertura por planos de saúde e discriminação na obtenção de emprego, trazendo conseqüências adversas, sociais e pessoais 34.
REFERÊNCIAS BIBLIOGRÁFICASl. POST, J. C. - Molecular biology in pediatric otolaryngology. In: Bluestone, C.D.; Stool, S.E.; Kenna, M.A. - Pediatric Otolaryngology, 3"' edition, Philadelphia, W.B. Saunders Company, 1996: 71-83.
2. DAVIS, A. C. - The prevamnce of hearing impairment and reported hearing disability among adults in Great Britain. IntJ. Epidemiol., 18: 911-917, 1989.
3. MORTON, N. E. - Genetic epidemiology of hearing impairment. Ann. N.Y. Acad. Sci., 630:16-31, 1991.
4. MARAZITA, M. L.; PLOUGHMAN, L. M.; RAWLINGS, B.; REMINGTON, E.; ARNOS, K. S.; NANCE, W. E. - Genetic epidemiological studies of early-onset deafness in the US schoolage population. Am. J. Med. Genet., 46 486-491, 1993.
5. LUBIANCA NETO, J. E.; LU, L.; EAVEY, R. D.; FLORES, M. A.; CALDERA, R. M.; SANGWATANAROJ, S.; et al. -The Bjornstad syndrome (sensorineural hearing loss and pili torti) disease gene maps to chromsome 2834-36. Am. J. Hum. Genet., 62: 11071112, 1998.
6. NADEAUJ. H.; KOSOWSKY, M.; STEEL, K. P. - Comparative gene mapping, genome duplication, and the genetics of hearing. Ann. NY. Acad. Sci., 630: 49-67, 1991.
7. STEEL, K. P. - Similarities between mice and human with hereditary deafness. Ann. N. Y. Acad. Sci., 630: 68-79, 1991.
8. PETIT, C. - Genes responsible for human hereditary deafness: symphony of a thousand. Nature Gener, 14: 385-391, 1996.
9. VÀN CÁMP, V.; WILLEMS, P. J.; SMITH, RJ. H. - Nonsyndromic hearing impairment: unparalleled heterogeneity. Am. J. Hum. Genet., 60: 758-764, 1997.
10. VAN CAMP, G.; SMITH, R. J. H - Hereditay Hearing Loss Homepage. World Wide Web URL: http://dnalab-www.uia.ac.be/dnalab/hhh, august 1998.
11. KIKUSHI, T.; KIMURA, R. S.; PAUL, D. L.; ADAMS, J. C. - Gap junctions in the rat cochlea: immunohistochemical and ultrastructural analysis. Anat.-Embríol., 191: 101-118, 1995.
12. KHETARPAL, U.; SCHUKNECHT, H. F.; GACEK, R. R.; HOLMES, L. B. -Autosomal dominant sensorineural hearing loss: pedigrees, audiologic and temporal bone findings in two kindreds. Arcb. Otolaryngol. Head Neck Surg., 117 1032-1042, 1991.
13. WANG, A.; LIANG, Y.; FRIDELL, R. A.; PROBST, F. J.; WILCOX, E. R.; TOUCHMAN J. W. et al - Association of unconventional myosin MY015 mutations with human nonsyndromic deafness DFNB3. Science, 280. 1447-1451, 1998.
14. PROBST, F. J.; FRIDELL, R. A.; RAPHAEL, Y.; SAUNDERS, T. L.; WANG, A.; LIANG, Y.; et al - Correction of deafness in shaker2 mice by na unconventional myosin in a BAC transgene. Science, 280: 1444-1447, 1998.
15. WEIL, D.; KUSSEL, P.; BLANCHAR, S.; LEVY, G.; LEVI-ACOBAS, F.; DRIRA, M.; et al -The autosomal recessive isolated deafness, DFNB2, and the Usher 113 syndrome are allelic defects of the myosin-VIIA gene. Nature Genet., 16..191~193, 1997.
16. LIU, X.Z.; WALSH, J.; TAMAGAWA, Y.; KITAMURA, K.; NISHIZAWA, M.; STEEL, K.; et al - Autosomal dominant non syndromic deafness (DFNAII) caused by a mutatiort in the Inyosin VIIA gene. Nature Genet., 17 268, 1997.
17. WOL, D.; BLANCHARD, S.; KAPLANj; GUILFORD, P.; GIBSON, F.; WALSH, J.; et al. - Defective myosin V1IA gene responsible for Usher syndrome rype 113. Nature, 374: 60-61, 1995.
18. SEU, T.; MAHONY, M.; FLEMING, J.; WALSH, J.; BROWN, D.; STEEL, K. P. - Shaker-1 mutations reveal roles for myosin V11A in both development and function of cochlear hair cells. Development, 125: 557-566, 1998.19. STEEL, K. P.; BROWN, S. D. -More deafness genes. Science, 280: 1403, 1998.
20. KELSELL, D. P.; DUNLOP, J.; STEVENS, H. P.; LENCH, N. J.; LIANG, J. N.; PARRY, G.; et al. - Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature, 387 80-83, 1997.
21. DENOYELLE, F.; WEIL, D.; MAW, M. A.; WILCOX, A. S.; LENCH, N. J.; ALLEN-POWELL, D. R.; et al. - Prelingual deafness: high prevalence of a 30deIG mutation in the connexin 26 gene. Hum. Mol. Genet., 6: 2173-2177. 1997.
22. DENOYELLE, F.; LINA-GRANADE, G.; PLAUCHU, H.; BRUZZONE, R.; CHAIB, H.; LEVI-ACOBAS, F.; et al. -Connexin 26 gene linked to a dominant deafness. Nature, 393:319-320,1998.
23. LYNCH, E. D.; LEE, M. K.; MORROW, J. E.; WELCSH, P. L.; LÉON, P. E.; KING, M. C. - Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous. Science, 278:1315-1318, 1997.
24. de KOK, Y .J.; VAN DER NIAAREL, S. M.; BITNER-GLINDZICZ, M.; HUBER, I.; MONACO, A. P; MALCOLM, S.; et al. -Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science, 267. 685-688, 1995.
25. VARAVA, O.; MORELL, R.; LYNCH, E. D.; WEISS, S.; KAGAN, M. E.; AHITUV, N., et al. - Mutation in transcription factor POU4F3
associated with inherited progressive hearing loss.in humans. Science, 279. 1950-1954, 1998.
26. VERHOEVEN, K.; VAN LÁER, L.; KIRSCHHOFER, K.; LEGAN, P. K.; HUGHES, D. C.; SCHATTEMAN, I.; et ai - Mutations in the human alfa-tectorin gene cause autosomal dominant nonsyndromc hearing impairment. Nature Genet., 19.-60-62, 1998.
27. EVERETT, L. A.; GLASER, B.; BECK, J. C.; IDOL, J. R.; BUCHS, A.; HEYMAN, M.; et al. -Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nature Genet., 17:411-422, 1997.
28. LI, X. C.; EVERETT, L. A.; LALWANI, A. K.; DESMUKH, D.; FRIEDMAN, T. B.; GREEN, E. D.; et al. - A mutation in PDS causes non-syndronlicrecessivedeafness. NatureGenet.,18: 215-217,1998.
29. HOLLWAY, G. E.; SUTHERS, G. K.; BASE, K. M.; TURNÉR, A. M.; DAVID, J. D.; MULLEY, J. C. - Deafness due to Pro250Arg mutation of FGFR3. Lancet, 351: 877-878, 1998.
30. ESTIVILL, X.; GOVEA, N.; BARCELO, A.; PERELLO, E.; BADENAS, C.; ROMERO, E.; et al. - Familial progessive sensorineural deafness is mainly due to the mtDNA A1555G mutation and is enhanced by treatment with aminoglycosides. Am. J. Hum. Genet., 62:27-35, 1998.
31. CHEN, A. H.; MUELLER, R. F.; PRASAD, S. D.; GREINWALD, J. H.; MANALIGODj; MUILENBURG, A. C., et ai. -Presymptomatic diagnosis of nonsyndromic hearing loss by genotyping. Arch. Otolaryngol.Head Neck Surg, 124:20-24,1998.
32. GHODSIAN, N. F. - Mitochondrial mutations and hearing loss: paradigm for mitochondrial genetics. Am j Hum. Genet., 62: 15-19, 1986.
33. GOLDSTEIN, D. A. - Ethical dilemmas in molecular genetics. Arch. Otolaryngol. Head Neck Surg., 119: 1183-1186, 1993.
34. WEINBERG, J. M. - Breaking bonds: discrimination in the genetic revolution. JAMA, 268. 1767, 1992.
*Professor Assistente e Regente da Disciplina de Otorrinolaringologia do Departamento de Oftalmo-Otorrinolaringologia da Faculdade Federal de Ciências Médicas de Porto Alegre. Chefe do Ambulatório do Serviço de Otorrinolaringologia Pediátrica do Hospital da Criança Santo Antônio, de Porto Alegre, e do Serviço de Otorrinolaringologia do Complexo Hospitalar Santa Casa, de Porto Alegre. Ex-Fellow da Divisão de Otorrinolaringologia Pediátrica do Massachusetts Eye and Ear Infirmary, Boston, EUA.
**Fellow do Departamento de Genética, Harvard Medical School, Boston, EUA. Pesquisador do Laboratório de Biologia Molecular do Instituto do Coração, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo.
Endereço para correspondência: Dr. José Faibes Lubianca Neto - Rua Mostardeiro, 333 - Conjt°. 3o6 - 90430-001 Porto Alegre /RS - Brasil. Telefone/fax: (051) 346.3831 - E-mail: iran@vanet.com.br Artigo recebido em 13 de outubro de 1998. Artigo aceito em 6 de abril de 1999.
