

Ano: 2001 Vol. 67 Ed. 6 - Novembro - Dezembro - (19º)
Seção: Relato de Caso
Páginas: 880 a 884
 PDF PT
PDF PT Hipogonadismo hipogonadotrófico e anosmia: síndrome de Kallmann
Hypogonadotropic hypogonadism and anosmia: Kallmann's syndrome
Autor(es):
Viviane Bom Schmidt 1,
Renato Roithmann 2,
Helena von Eye Corleta (3,4,6),
Edison Capp (3,4,5)
Palavras-chave: síndrome de Kallmann, hipogonadismo hipogonadotrófico, anosmia
Keywords: Kallmann's syndrome, hypogonadism hypogonadotrophic, anosmia
Resumo:
A síndrome de Kallmann caracteriza-se pela associação de hipogonadismo hipogonadotrófico à anosmia ou hiposmia. É causada por um defeito na migração dos neurônios que produzem o GnRH e dos neurônios que formam os nervos olfatórios. A doença afeta somente a secreção de gonadotrofina, sendo que todos os outros hormônios hipofisários são secretados normalmente. Neste trabalho são relatados dois casos de síndrome de Kallmann e apresentada revisão da literatura.
Abstract:
Kallmann's syndrome is characterized by hypogonadotrophic hypogonadism and anosmia or hyposmia. Its primary defect is an anomalous migration of the neurons responsible for the GnRH production and the neurons that compose the olfactory nerves. The syndrome affects the gonadotrophin production only, all other hypophysial hormones are normally secreted. In this paper we report and discuss two cases of Kallmann's syndrome in young women.
![]()
INTRODUÇÃO
A síndrome de Kallmann caracteriza-se pela associação de hipogonadismo hipogonadotrófico à anosmia ou hiposmia2. O traço é transmitido geneticamente de forma recessiva ligada ao X ou como um caráter autossômico dominante limitado ao sexo masculino, podendo ocorrer a heterogeneidade genética. É causada por um defeito na migração dos neurônios que produzem o hormônio de liberação de gonadotrofinas (GnRH) e dos neurônios que formam os nervos olfatórios.
A síndrome afeta somente a secreção de gonadotrofina, sendo que todos os outros hormônios hipofisários seguem secretados normalmente. Assim, quando administra-se GnRH há aumento de hormônio luteinizante (LH) e hormônio folículo estimulante (FSH), indicando que o defeito básico pode ser no hipotálamo (com síntese insuficiente de GnRH), ou um defeito na neurotransmissão de GnRH (resultando em síntese inadequada), ou ambos.
Neste trabalho são relatados dois casos de síndrome de Kallmann e apresentado revisão da literatura.
APRESENTAÇÃO DE CASOS CLÍNICOS
CASO 1
ILS, feminina, 20 anos, consultou por infertilidade. Na história relatava amenorréia primária e uso de anticoncepcional oral desde os 18 anos de idade. Na revisão dos sistemas referia anosmia. Ao exame físico paciente com altura de 159 cm, pesando 52 kg. Mamas em estágio M3 e pêlos em estágio P2 segundo os critérios de Tanner. A vagina estava trófica (em uso de anticoncepcional oral), restante do exame físico sem particularidades. A hormonioterapia foi suspensa e foram solicitados os seguintes exames: hormônio folículo estimulante (FSH) < 0,38 mU/ml, hormônio luteinizante (LH) 0,7 mU/ml, prolactina 11 ng/ml. A ecografia transvaginal mostrou útero (6x5x4cm), endométrio (0,3cm), ovário direito (2x1,5x1cm) e ovário esquerdo (2,1x1x1,3cm). Foi realizado o teste de estímulo hipofisário com 100µg de GnRH. A dosagem de LH basal foi de 0,7 mU/ml e a de FSH 1,3 mU/ml; 30 min após estímulo com hormônio de liberação de gonadotropinas (GnRH) o LH foi de 73 mU/ml e o FSH 40,4 mU/ml. A tomografia computadorizada mostrou ausência de lesões expansivas do sistema nervoso central e da hipófise. Pela tomografia computadorizada também descartou-se lesões no teto da cavidade nasal ou seios paranasais. Diante do quadro clínico e dos resultados apresentados pelos exames foi diagnosticado síndrome de Kallmann. Nessa ocasião, pelo desejo de gestar, foi induzida ovulação com gonadotrofina sob controle ecográfico.
CASO 2
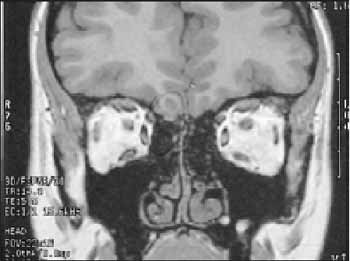
RS, feminina, estudante. Com 21 anos paciente procurou orientação otorrinolaringológica devido à anosmia. A história clínica não revelou outras queixas otorrinolaringológicas. Endoscopia nasosinusal e tomografia de seios paranasais sem alterações. O otorrinolaringologista a encaminhou ao ginecologista, pois a paciente estava preocupada porque seu ginecologista afirmara que não poderia gestar, pois, após vários exames, havia sido constatada disgenesia gonadal. Aos 21 anos de idade, paciente veio à consulta ginecológica com diagnóstico de disgenesia gonadal. Relatava uso de anticoncepcional oral desde os 17 anos para menstruar. Após uso de anticoncepcional oral iniciou desenvolvimento de mamas e alguns pêlos, entretanto não estava satisfeita com o grau de desenvolvimento de seus caracteres sexuais secundários. Ao exame físico a pressão arterial era 110/60 mmHg, estatura 160cm, peso 58,6kg, pêlos axilares e pubianos diminuídos, restante do exame sem alterações. Trazia consigo os seguintes exames: LH < 0,5 mU/ml, FSH 1,5 mU/ml, prolactina 4,8 ng/ml, cariótipo (46XX) e ressonância magnética do cérebro que mostrava ausência de bulbo e trato olfatório à esquerda; à direita essas estruturas estavam presentes (Figura 1). Trazia ainda teste do GnRH com resultado de FSH > 200 mU/ml e de LH > 150 mU/ml uma hora após estímulo. Neste momento, após interpretação dos exames foi diagnosticado síndrome de Kallmann, sendo receitado estrogênio natural e progestágeno cíclico. A paciente foi orientada quanto ao diagnóstico que, diferentemente da disgenesia gonadal, é compatível com a gestação, visto ser o problema hipotalâmico e não ovariano.
Figura 1. Corte coronal de RM cm mostrando ausência de bulbo e trato olfatório à esquerda.
DISCUSSÃO
A Síndrome de Kallmann (SK), com prevalência estimada em 1/10.000 homens e 1/50.000 mulheres9, é caracterizada pela associação de hipogonadismo hipogonadotrófico e anosmia ou hiposmia. Há suposições de que a anosmia ocorra devido à ausência dos nervos olfatórios, bulbos, e sulco - arrinencefalia9, conseqüência de uma falha na migração neuronal, envolvendo o hormônio GnRH e os nervos olfatórios, para o sistema nervoso central¹². Estudos imunohistoquímicos mostraram que os neurônios responsáveis pela secreção de GnRH migram da placa olfatória para o hipotálamo precocemente na embriogênese8. A migração dos neurônios responsáveis pela secreção de GnRH e dos nervos olfatórios ocorre dentro dos limites das meninges, acima da lâmina cribiforme7. Assim, não ocorrendo essa migração o hipogonadismo hipogonadotrófico ocorre.
O traço é transmitido geneticamente de forma recessiva ligada ao X ou como um caráter autossômico dominante, podendo ocorrer a heterogeneidade genética¹.
O gene KAL, no braço curto do cromossoma X (Xp22.3), codifica uma proteína (anosmin-1) responsável pelas funções necessárias para a migração neuronal. Esse gene é o responsável pela forma ligada ao X da doença3,4,5.
A avaliação dos pacientes com deficiência de gonadotrofina exige uma história familiar meticulosa para identificar outros membros da família com essa síndrome, já que alguns integrantes da família podem apresentar anosmia ou hiposmia, sem qualquer disfunção reprodutiva.
É caracterizada pela deficiência de gonadotrofina e por hipogonadismo, com conseqüente falência da função gametogênica e da produção sexual de esteróide. Infantilismo sexual é a manifestação mais saliente da síndrome, visto que o desenvolvimento de características sexuais secundárias é dependente de esteróides sexuais. O grau de hipogonadismo em pacientes masculinos varia desde completa imaturidade testicular e células de Leydig atrofiadas à hipoleidigismo (caracterizada por volume testicular e função espermática quase normais).
Em mulheres, as características clínicas variam de achados eunucóides clássicos a desenvolvimento moderado das mamas. Amenorréia primária, imaturidade sexual, níveis abaixo do normal de gonadotrofinas, cariótipo feminino normal e anosmia ou hiposmia são achados clínicos. Os ovários dessas pacientes raramente contêm folículos que passaram do primeiro estágio de desenvolvimento, sugerindo que os estágios recentes da maturação folicular exigem uma quantia de gonadotrofina que está além daquela secretada por essas pacientes. A imaturidade ovariana responde prontamente à gonadotrofina exógena ou a estimulação pulsátil com GnRH, assim é possível que ocorra ovulação e, até mesmo, gestações.
A doença afeta somente a secreção de gonadotrofina, e todos os outros hormônios hipofisários são secretados normalmente. Os níveis circulantes de LH eFSH são usualmente detectáveis, todavia os valores encontrados são bem inferiores aos considerados normais. Caso haja estimulação exógena com GnRH, a maioria dos pacientes aumenta os níveis circulantes de LH e FSH. Ressalta-se que os graus de resposta à estimulação com GnRH podem variar desde falência do LH e FSH em aumentar seus níveis circulantes, aumento apropriado de LH e FSH, e aumento apenas de LH ou apenas de FSH. As pacientes relatadas neste trabalho tiveram aumento tanto do LH quanto do FSH após estímulo com GnRH. Uma das causas de resposta inadequada ao teste do GnRH é o hipoestrogenismo das pacientes. A hipófise quando em ambiente hipoestrogênico não responde com o aumento esperado das gonadotrofinas. Nossos dois casos responderam adequadamente provavelmente por haverem usado há pouco tempo anticoncepcional oral.
Os pacientes podem apresentar, além das características clínicas acima citadas, defeitos na linha média, lábio leporino, fenda palatina, agenesia renal, surdez sensorioneural9, infertilidade, paraplegia espástica, disfunção cerebelar e nistagmo10.
O diagnóstico de anosmia pode ser obtido através de uma anamnese minuciosa, um exame físico detalhado, incluindo endoscopia dos terços médio e superior da cavidade nasal e exames complementares (testes olfatórios e imagem). A Tabela I resume o roteiro diagnósticocomumente empregado na avaliação de pacientes com alterações do olfato. Entre os testes olfatórios, o mais utilizado tem sido o teste de identificação de cheiro da Universidade da Pensilvânia (UPSIT)¹². O mesmo é composto por 20 itens que a pessoa deve cheirar em cada narina isoladamente. A endoscopia nasosinusal é igualmente importante para excluir causas comuns de diminuição do olfato, como obstrução do fluxo aéreo por pólipos ou processos inflamatórios do nariz e seios paranasais (rinossinusites). Os testes de imagem incluem a tomografia computadorizada (TC) e a ressonância magnética (RM). A ressonância magnética é indicada caso não se descubra a causa da anosmia com os exames anteriores, incluindo a TC. A RM do encéfalo deve enfatizar o hipotálamo-hipófise, para excluir existência de uma massa encefálica. Não sendo descartada SK, deve-se enfatizar, na RM, o bulbo e o trato olfatório, nesse caso, estudo genético e endocrinológico são imprescindíveis. É importante enfatizar que um critério para o diagnóstico da SK deve incluir, além de anosmia, a demonstração de anormalidades no bulbo olfatório12.
A TC não é indicada para diagnosticar ausência ou presença do aparato olfatório, uma vez que sua visualização é limitada com a TC, devido à existência de artefatos na base do crânio. Assim, a RM é o melhor método para o estudo do bulbo e trato olfatório, já que as imagens não são afetadas pelos artefatos da base do crânio. Além disso, pode detectar outras causas de disfunção olfatória, incluindo meningiomas ao longo da lâmina cribiforme, injúria pós-traumática, coleções extra-axiais, e outras massas nasosinusais que podem afetar a função olfatória10. O sistema olfatório é melhor analisado por ambas seqüências de cortes da RM: T1, para análise volumétrica; e T2, para identificação de áreas do sistema olfatório danificadas.
O diagnóstico diferencial da SK deve ser feito com infecção viral, infecção intracraniana (meningite e encefalite), injúria pós-traumática ao epitélio olfatório, atresia de coana e hipogonadismo hipogonadotrófico idiopático (Tabela 2). Ressalta-se que a única diferença clínica entre pacientes com SK e hipogonadismo hipogonadotrófico idiopático é a anosmia, presente nos pacientes com SK.
O manejo com os pacientes acometidos por essa síndrome é precário. Há pouco a oferecer às pessoas com hiposmia ou anosmia congênita. É importante ressaltar que, muitas vezes, as pessoas não estão cientes de seu defeito olfatório. Por essa razão, deve-se alertar os pacientes quanto a ambientes que dependem do olfato, evitando contato com produtos químicos arriscados e gases12. As imagens obtidas com a RM são importantes, uma vez que podem definitivamente responder às dúvidas referentes ao possível restabelecimento do sentido do olfato. Isso porque mostra a ausência ou hipoplasia do bulbo e trato olfatório, situação em que a restauração da função olfatória é limitada.
Entretanto a amenorréia e o desenvolvimento de caracteres sexuais secundários e a infertilidade podem ser satisfatoriamente tratados. O uso de estrogênios naturais inicialmente isolados permite inicialmente o desenvolvimento mamário e após associado a progestágeno cíclico a regularidade mesntrual. Quando o diagnóstico é retardado podemos encontrar pacientes com hábito eunucóide pelo não fechamento das epífises. O prognóstico quanto à gestação é satisfatório. A ovulação pode ser induzida com uso de análogos do GnRH de forma pulsátil ou diretamente com o uso de gonadotrofinas.
Pacientes com Síndrome de Kallmann podem consultar inicialmente com o ginecologista ou com o otorrinolaringologista. É necessário que estes especialistas conheçam o quadro clínico da doença para que as pacientes tenham orientação e tratamento interdisciplinar adequados.
Tabela 1. Avaliação do paciente com alterações do olfato6.
1. História clínica detalhada:
Capacidade olfatória antes de sua perda
Hiposensação ou distorção da percepção
Possível associação com desordens gustatórias
Doença nasosinusal e tratamentos realizados
Uso de drogas e exposição tóxica
Trauma cefálico
Infecções do trato respiratório superior
Problemas neurológicos
Doenças metabólicas e endócrinas
Problemas psicológicos
Doença neoplásica
História familiar
2. Exame Físico:
Nariz - endoscopia nasosinusal pós-descongestionante tópico
Cabeça e pescoço
Exame neurológico
3. Testes olfatórios:
Teste de detecção
Teste de identificação
Potencial evocado
Avaliação do paladar
4. Imagem:
Tomografia computadorizada, Ressonância nuclear magnética
5. Estudos do fluxo nasal e patência - rinomanometria e rinometria acústica
6. Testes complementares:
Testes laboratoriais: hemograma, função hepática, função renal, avaliação metabólica e endócrina, etc.
Biópsia da mucosa olfatória
Tabela 2. Causas de disfunção olfatória6.Obs:listagem completa vide referência 6.
Lesões do nariz/ via aérea
Anormalidades estruturais (desvio de septo, adenóides, outras)
Pólipo Nasal
Rinites Alérgica e Não-Alérgicas
Nutrição/metabolismo
Deficiência de Vitamina
Vitamina A
Vitamina B6
Vitamina B12
Deficiência de Metais
Zinco
Cobre
Má-nutrição proteico-calórica
Nutrição Parenteral Total (sem reposição adequada)
Fibrose Cística
Falência Renal Crônica
Cirrose Hepática
Gota
Doença de Whipple
Tumores
Intracranianos
Intranasais (estesioneuroblastoma, outros)
Sistêmicos (pulmão, trato gastrointestinal, etc.)
Neurológicas
Esclerose Amiotrófica Lateral
Disautonomia Familiar
Síndrome de Refsum
Esclerose Múltipla
Doença de Parkinson
Paralisia Supranuclear Progressiva
Epilepsia do Lobo Temporal
Esclerose Mesial Temporal
Hamartomas
Infarto Prévio
Miastenia Grave
Retinite Pigmentosa
Insuficiência Vascular e Anoxia
Pequenos Acidentes Vasculares Múltiplos
Ataque Isquêmico Transitório
Síndrome do "roubo" da Subclávia
Outros
Abscesso Cerebral (região frontal ou etmoidal)
Meningite
Sífilis
Siringomiela
Doença de Paget
Doença de Korsakoff
Hidrocefalia
Enxaqueca
Endócrinas
Insuficiência Adrenal Crônica - Doença de Addison
Hiperplasia Congênita Adrenal
Síndrome de Cushing
Hipotireoidismo
Diabetes Mellitus
Amenorréia Primária
Disgenesia Gonadal da Cromatina Negativa - Síndrome de Turner
Hipogonadismo Hipogonadotrófico - Síndrome de Kallmann
Hipogonadismo Hipergonadotrófico
Pseudohipoparatireoidismo
Panhipopituitarismo
Gigantismo
Distrofia Adiposogenital - Síndrome de Froelich
Congênita/Hereditária
Síndrome de Hipogeusia e Hiposmia
Tr de:
Fenda Submucosa do Palato Duro
Hipoplasia Facial
Crescimento Atrofiado
"Doença Red-Haired" com Alteração Pigmentar
Anosmia de Origem Genética Completa e Específica
Asma Brônquica
Hipertelorismo Orbitário
Traumatismo craniano e/ou nasal
Drogas
Esteróide Adrenal (uso crônico)
Excesso de Aminoácidos
Histidine
Cisteine
Anestésico, Locais
Procaína HCl
Cacaína HCl
Tetracaína HCl
Agentes Anticancerígenos (metotrexate)
Anti-histamínicos (maleato de clorfeniramine)
Antimicrobianos
Griseofulvina
Lincomicina
Streptomicina
Tetraciclina
Tirotricina Intranasal
Neomicina Local
Neoarsfenamina
Antireumáticos
Mercúrio ou Sais de Ouro
D-Penicilamine
Antitireóide
Metimazole
Propiltiouracil
Tiouracil
Medicação com Hiperlipoproteinemia
Clofibrato
Colestiramina
Soluções Intranasais Salinas com:
Acetilcolina
Acetil, â-metilcolina
Mentol
Stricnina
Sulfato de Zinco
Opiáceos
Codeína
Hidromofone HCl
Morfina
Psicofarmacêuticos (psilocibin, LSD)
Simpaticomiméticos
Sulfato de Anfetamina
Teoclate de Fenmetrazina
Fenbutrzate HCl
Outros
Antipirina
ETOH Oral
Vasoconstritores Locais
Cimetidina
L-dopa
Poluentes Químicos (gasosos e poeira química)
Intervenção Médica
Cirurgia intracraniana e/ou nasal
Radioterapia
Outros
Psiquiátrica
Esquizofrenia
Síndrome de Referência Olfatória
Desordens Depressivas
Histeria
Outros
Presbiosmia
Processos Fisiológicos
Variação Circadiana
Período menstrual
Gravidez
Idiopática
REFERÊNCIAS BIBLIOGRÁFICAS
1. BICK, D.P.; BALLABIO, A. - Bringing Kallmann's syndrome into focus. AJNR, 14:852-4, 1993.
2. KALLMANN, F.J.; SCHONFELD, W.A.; BARERRA, S.E. - The genetic aspects of primary eunuchoidism. Am. J. Ment. Defic., 48:203-36, 1944.
3. MAYA-NUÑEZ, G.; TORRES, L.; ULLOA-AGUIRRE, A.; ZENTENO, J.C.; CUEVAS-COVARRUBIAS, S. et al. - An atypical contiguous gene syndrome: molecular studies in a family with X-linked Kallmann's syndrome and X-linked ichthyosis. Clinical Endocrinology, 50:157 62, 1999.
4. MEITINGER, T.; HEYE, B.; PETITE, C. et al. - Definite localization of x-linked Kallmann syndrome to Xp22.3: close linkage to the hypervariable repeat sequence CRI-S232. Am J Hum Genet, 47:664-660, 1990.
5. PETIT, C.; LEVILLIERS, J.; WEISSENBACH, J. - Long-range restriction map of the terminal part of the human X chromosome. Iproc Natl Acad Sci USA, 87:3680-4, 1990.
6. ROITHMANN, R.; ROTH, Y.; COLE, P.; CHAPNIK, J.; HYDE, M. - Anosmia - in Diagnosis and Treatment of Symptoms of the Respiratory Tract. Armonk, New York, Ed. Irwin R, Curley F, Grossman R, 557 80, 1997.
7. RUGARLI, E.I.; GHEZZI, C.; VALSECCHI, V. & BALLABIO, A. - The Kallmann syndrome gene product expressed in COS cells is cleaved on the cell surface to yield a diffusible component. Human Molecular Genetics, 5:1109-15, 1996.
8. SCHWANZL-FUKUDAM.; BICK, D.; PFAFF, D.W. - Luteinizing hormone-releasing hormone (LH-RH) - expressing cells do not migrate normally in na inherited hypogonadal (Kallmann) syndrome. Mol Brain Res, 6:311-26, 1986.
9. VOGL, T.J.; STEMMLER, J.; HEYE, B.; SCHOPOHL, J.; DANEK, A.; BERGMAN, C.; BALZER, J.O; FELIX, R. - Kallman syndrome versus idiopathic hypogonadotropic hypogonadism at MR imaging. Head and Neck Radiology, 191:53-7, 1994.
10. YOUSEM, D.M.; TURNER, W.J.D.; LI, C.; SNYDER, P.J.; DOTY, R.L. - Kallmann syndrome: MR evaluation of olfatory system. AJNR, 14:839 43, 1993.
11. YOUSEM, D.M.; GECKLE, R.J.; BILKER, W.; MCKEOWN, D.A.; DOTY, R.L. - MR evaluation of patients with congenital hyposmia or anosmia. AJR, 166:439-43, 1996.
WORTSMAN, J.; HUGHES, L.F. - Case report: olfactory function in a fertile eunuch with Kallmann Syndrome. Am J Med Sci, 311(3):135-8, 1996.
1 Acadêmica da Universidade Luterana do Brasil, Canoas, Rio Grande do Sul e Monitora da Disciplina de Otorrinolaringologia.
2 Professor Adjunto de Otorrinolaringologia, Universidade Luterana do Brasil, Canoas, Rio Grande do Sul.
3 Serviço de Ginecologia do Hospital de Clínicas de Porto Alegre.
4 Departamento de Ginecologia e Obstetrícia da Universidade Federal do Rio Grande do Sul, Porto Alegre.
5 Departamento de Fisiologia da Universidade Federal do Rio Grande do Sul.
6Gerar - Centro de Fertilização Assistida.
Trabalho realizado na Universidade Luterana do Brasil.
Endereço para correspondência: Viviane Bom Schmidt - Travessa Fortaleza, 115 - 91720-500 Porto Alegre, RS, Brasil.
Telefone: (0xx51) 266-1068 - E-mail: vivibs@pro.via-rs.com.br
Artigo recebido em 10 de maio de 2001. Artigo aceito em 26 de junho de 2001.
Imprimir: ![]()