

Ano: 2001 Vol. 67 Ed. 6 - Novembro - Dezembro - (18º)
Seção: Relato de Caso
Páginas: 873 a 878
 PDF PT
PDF PT Angiofibroma juvenil com evolução atípica
Atypical evolution of juvenile nasopharyngeal angiofibroma
Autor(es):
Dr. Lídio Granato 1,
Dr. Inácio Koji Hashimoto 2
Palavras-chave: angiofibroma juvenil, extensão intracraniana, tratamento cirúrgico, radioterapia
Keywords: juvenile nasopharyngeal angiofibroma, intracranial extension, radiation therapy
Resumo:
Paciente com 34 anos de idade apresentou angiofibroma juvenil com evolução atípica. A história do paciente era de 14 anos de evolução e o tumor ocupava os espaços extra e intracranial em grande extensão. Submetido à intervenção extracranial, foi em seguida encaminhado para radioterapia. O paciente desenvolveu um nódulo na face, no local da incisão externa, utilizada na intervenção, com características de benignidade. A massa recidivou por mais duas vezes, sempre na mesma localização e com os mesmos aspectos histológicos, porém com recidivas cada vez mais próximas e mais sangrantes. Embora a avaliação histológica mostrasse característica de benignidade, o comportamento clínico era de malignidade. O paciente teve seu estado geral agravado e faleceu seis meses após a radioterapia.
Abstract:
The authors report a 34-year-old patient presenting with an atypical juvenile angiofibroma. Symptoms had begun 14 years prior to diagnosis and the tumor occupied large extra and intracranial spaces. Following the last surgical intervention, the patient underwent radiotherapy and developed a nodule with benign characteristics on the face at the site of the incision. The mass was removed and recurred twice at the same site with the same histological characteristics. Recurrences occurred closer together and became progressively more hemorrhagic. Although histological characteristics remained benign, the clinical presentation suggested malignancy. The patient presented clinical complications and died six months after radiation therapy.
![]()
Introdução
O angiofibroma juvenil é um tumor benigno, raro, que ocorre numa freqüência de 0,5% de todas as neoplasias da cabeça e pescoço3. O crescimento do angiofibroma juvenil possibilita a sua extensão intracraniana, e de acordo com vários autores varia de 4,3 a 11%4,5,2,11,13. Maiores incidências são relatadas por Gupta e colaboradores com 33% e Bremer e colaboradores com 26%.
O acompanhamento de muitos pacientes portadores de angiofibroma juvenil na Santa Casa de São Paulo nos deu a convicção de que a evolução do nosso paciente foi atípica. A literatura não mostra situação semelhante e por esta razão julgamos interessante divulgar o caso.
Apresentação do Caso Clínico
A.F.B.S. - 34 anos, branco, comerciário, procedente do Maranhão. Data de admissão 03/11/97.
HMA - Paciente refere sangramento nasal há 14 anos. Cinco anos após o início do quadro, foi submetido à biópsia que resultou no diagnóstico de angiofibroma juvenil. Quatro anos depois, foi submetido à cirurgia na sua cidade. Permaneceu relativamente bem por cinco anos, porém esporadicamente apresentava sangramentos nasais, que eram discretos.
Há oito meses, houve agravamento das epistaxes e, concomitantemente, surgiu redução da acuidade visual do olho direito.
Há dois meses vem apresentando obstrução nasal direita e logo em seguida cefaléia fronto-parietal D. Diariamente também apresenta sensação de ouvidos "tapados".
Procurou outro facultativo na sua cidade que o submeteu a nova biópsia, que confirmou o diagnóstico anterior. Solicitada a tomografia computadorizada, foi constatada uma grande extensão do tumor intracrânio e encaminhado para nosso Serviço.
Exame ORL
No exame otorrinolaringológico, observou-se na inspeção ptose palpebral direita e anisocoria com midriase a D. com redução do reflexo pupilar.
Cavidade do nariz - tumoração com coloração avermelhada com crostas hemáticas ocupando a profundidade da fossa nasal D.
Cavidade oral/Orofaringe - fístula oro-nasal de mais ou menos um centímetro de diâmetro, na transição palato duro e mole.
Rinofaringe - ocupada inteiramente por massa tumoral maior do lado direito.
Paciente foi internado logo após o exame por apresentar epistaxe com repercussão hemodinâmica. Foi colocado tampão tipo "dedo de luva" e foram administrados estrogênios conjugados, E.V.(20 mg) - 1 amp./dia, por poucos dias. A seguir, passou a receber 1 comprimido (0,625 mg) via oral, até cessar o sangramento.
A avaliação hematológica mostrava hemoglobina igual a 10 g/dl.
Solicitada nova tomografia computadorizada e ressonância nuclear magnética, a tomografia computadorizada revelou: desvio de septo para a esquerda e tumor que provocava erosão das lâminas pterigóides à direita, da parede lateral direita do seio esfenoidal, células etmoidais e rinofaringe, estendendo-se para fossa média. Tal formação ainda desvia a parede medial do seio maxilar direito, que se apresenta ocupado por líquido espesso. Erosão do palato duro e assoalho da órbita direita.
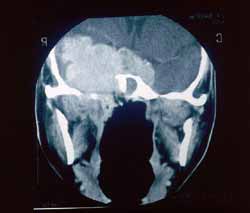
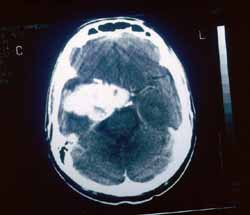
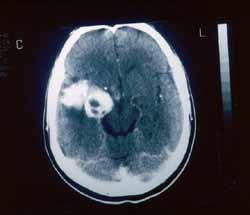
A invasão intracraniana mostra lesão expansiva lobulada que apresenta intenso realce determinando alargamento do forame oval, invasão do seio cavernoso, englobando e deslocando superior e lateralmente a porção intracavernosa da artéria carótida interna (Fig. 1). A lesão em corte axial com contraste na fossa média envolve o seio esfenoidal e adere a duramáter do ligamento petro-clinoideo (Fig. 2). O tumor alcançou a altura do terceiro ventrículo (Fig. 3).
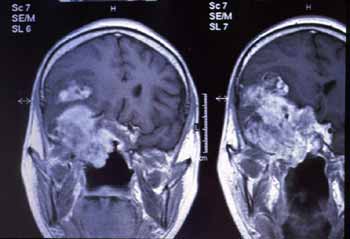
A ressonância magnética confirmou os mesmos achados da tomografia computadorizada (Fig. 4).
Figura 1. Corte coronaL, mostrando lesão expansiva lobulada que apresenta intenso realce, localizada na rinofaringe à direita com invasão de seio cavernoso.
Figura 2. Corte axial com contraste: lesão ocupando a fossa média com envolvimento do seio esfenoidal e aderido à duramáter do ligamento petro clinoideo D.
Figura 3. Lesão alçando a altura do 3º ventrículo.
Figura 4. Ressonância: Corte coronal em T1 com contraste mostrando lesão invadindo base do crânio.
Na evolução o paciente continuou com a ptose palpebral (superior), porém, houve agravamento da visão com a amaurose a D., redução da mobilidade do globo para abduzir (lesão do VI par) e logo em seguida fixação do globo por lesão completa do III par (óculomotor).
Também o olho esquerdo apresentou diminuição da acuidade visual.
Indicada a cirurgia, com a finalidade de remoção extracranial do tumor. Na véspera da cirurgia o paciente necessitou transfusão de uma unidade de concentrado de hemácia, pois apresentava hemoglobina de 8,3 g/dl.
Submetido à cirurgia por acesso de rinotomia lateral e técnica transantral direita. Feita a ressecção do tumor que ocupava a fossa nasal D, rinofaringe, seio esfenoidal, células etmoidais e fossa pterigopalatina. A dissecção tumoral cessou na base do crânio, junto à fossa média craniana.
No pós-operatório imediato, o paciente apresentou um período de agitação que melhorou com o uso de corticóide sistêmico.
Três meses após a cirurgia foi novamente avaliado pela neurocirurgia, que contra indicou a remoção do extenso tumor, que ocupava o cérebro e foi aconselhado tratamento radioterápico. Após dois meses do tratamento observou-se que o paciente ainda continuava com desconforto, com muita cefaléia e piora da visão do lado esquerdo. O olho direito estava amaurótico e o olho esquerdo enxergava apenas vulto. Na tomografia computadorizada de controle houve pequena redução da massa intracraniana.
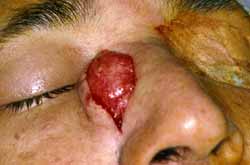
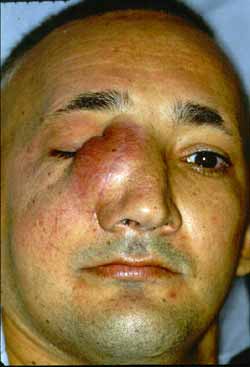
Na face surgiu nódulo no local da incisão da rinotomia lateral. Foi ressecado e encaminhado para anatomopatológico, que revelou angiofibroma juvenil (Fig. 5) Depois de um mês desta cirurgia surgiu no mesmo local novo nódulo, com as mesmas características (Fig. 6). O anatomopatológico confirmou o mesmo resultado. Após quatro meses o paciente nos procurou com a mesma queixa anterior, porém com uma tumoração facial, sempre na mesma localização, porém muito maior. A retirada desta massa expôs o osso próprio nasal, onde já havia erosão parcial do mesmo.
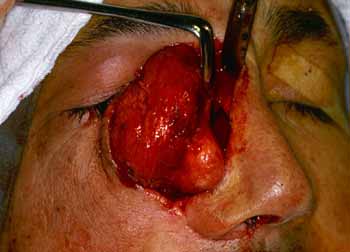
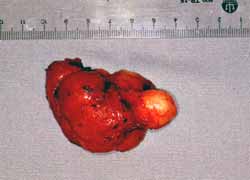
A lesão guardava sempre o mesmo aspecto, bem delimitada, dura e que diferenciava da anterior pela maior vascularização do tumor (Fig. 7 e 8). Paciente evoluiu com redução do nível de consciência e muita sonolência. Retornou à sua cidade de origem onde faleceu após seis meses.
Figura 5. Primeiro nódulo no local da incisão da rinotomia lateral.
Figura 6. Nódulo recidivado no mesmo local anterior.
Figura 7. Exérese do nódulo (angiofibroma).
Figura 8. Peça cirúrgica.
DISCUSSÃO
O angiofibroma juvenil é um tumor histologicamente benigno, porém clinicamente agressivo, apresentando-se localmente invasivo para estruturas vizinhas e muitas vezes comprometendo a base do crânio com invasão intracranial.
Acomete geralmente pacientes jovens do sexo masculino.
O tumor geralmente tem origem na porção póstero-nasal, bem junto do orifício esfenopalatino. Deste ponto ele pode se espalhar lateralmente para a fossa pterigopalatina, fossa infratemporal e algumas para a fossa temporal. O tumor pode se desenvolver também para cima através da fissura orbitária inferior, então caminha para trás em direção ao ápice da órbita e fissura orbitária superior para finalmente alcançar o seio cavernoso (invasão anterior)10,2.
Segundo os mesmos autores, quando o tumor progride posteriormente, pode erodir a lâmina medial do processo pterigóide, seguindo pelo assoalho do seio esfenoidal. Cresce dentro do seio ou se dirige para baixo para a fossa pterigóide, assim como para o espaço parafaríngeo. A massa tumoral pode se estender através do tecido ósseo da raiz do processo pterigóide para o corpo do esfenóide em direção ao clivus e à região medial do seio cavernoso (invasão medial). A chamada invasão inferior corresponde ao crescimento do tumor para inferior quando alcança o forame lácero, a artéria carótida interna e mesmo o seio cavernoso. Quando a tuba timpânica é comprometida, daqui o tumor pode alcançar o forame redondo e oval; deste ponto poderá haver extensão para o seio cavernoso - invasão lateral.
Estes tumores são sempre estudados pela tomografia computadorizada e muitas vezes pela ressonância magnética. Baseados nestes estudos, eles são classificadosem estádios. Entre muitas classificações, a mais simples é a de Chandler de 1984, que considera quatro estádios.
Andrews (1989) propôs modificação que subdivide o IV estádio, que seria aquele do tumor com invasão da cavidade intracraniana, em extradural e intradural. Esta distinção é particularmente importante porque estes tumores envolvem grandes complicações intra-operatórias, como: hemorragias, fístulas liquóricas, problemas oftalmológicos, assim como a grande possibilidade de recidiva (Gupta & Murthy).
Fagan e colaboradores apresentaram em 1997 trabalho com 16 casos, onde três pacientes apresentavam invasão intracranial de fossa média. Nesta casuística arecorrência foi de 37,5%, e os tumores que recidivaram se estenderam além dos ossos, comprometendo partes moles, tais como a fossa infratemporal, seio cavernoso e cérebro.
Herman e colaboradores, em 1999, apresentaram um trabalho contando com 44 pacientes em 1999, seguidos por um longo período pós-operatório. Eles concluíram que 2/3 dos tumores tinham invasão de base de crânio e a recorrência foi de 27,5%.
Os autores mencionam ainda que tumores que invadem a base do crânio comprometem o ápice da órbita ou a face anterior do seio cavernoso, e mesmo uma cirurgia cuidadosa não impede a oftalmoplegia e o decréscimo da acuidade visual.
Também a invasão do forame lácero leva sempre um perigo potencial em comprometer a artéria carótida interna.
O paciente procurou o Serviço de Otorrinolaringologia com idade avançada em relação à média dos pacientes portadores de angiofibroma. Este fato chamava a atenção, além do comprometimento ocular que foi se agravando paulatinamente. Na avaliação tomográfica, com contraste iodado, o paciente apresentava extensa formação expansiva lobulada que ocupava a rinofaringe e com grande extensão intracraniana, determinando alargamento do forame oval, invasão do seio cavernoso, englobando e deslocando superior e lateralmente a porção intracavernosa da artéria carótida interna à direita, com erosão do corpo do esfenóide adjacente à lesão.
A decisão de efetuar uma cirurgia extracranial surgiu após a contra indicação da neurocirurgia de tentar ressecar a massa intracraniana. O paciente continuava apresentando sangramentos nasais com muita freqüência e logo após a exérese do tumor, principalmente na sua porção nasal e rinofaringe, houve uma sensível melhora do quadro obstrutivo e da hemorragia.
Baseado na literatura, temos trabalhos de Wiatrak e colaboradores (1993) que publicaram três casos e Mcgahan e colaboradores (1989), 10 casos com pacientes submetidos à radioterapia, todos com extensão intracraniana, evoluindo com significante resolução da sintomatologia, porém com persistência do tumor, embora com redução de seu tamanho, comprovado com tomografia computadorizada.
Em função destes trabalhos e na impossibilidade de submeter o paciente à cirurgia, foi indicada a radioterapia. A utilização deste tratamento aparentemente não trouxe nenhum benefício para o paciente, pelo contrário, pois o mesmo passou a apresentar cefaléia muito intensa, pelo provável edema que se instalou.
Além disso, surgiu na linha de incisão um nódulo, bem superficial na pele, o qual após ser retirado e encaminhado a exame anatomopatológico, revelou tratar-se de angiofibroma. Esta massa surgiu mais duas vezes no mesmo local.
Este aparecimento foi inusitado e com certeza foi predisposto pela radioterapia. É muito raro o aparecimento de massa tumoral exteriorizando na pele, por provável implantação de células na linha de incisão. O que também chamou muito a atenção foi a recidiva tão precoce nas três oportunidades e sempre mantendo o padrão histológico de benignidade.
O que se pergunta nesta eventualidade é por que um tumor benigno passa a apresentar um comportamento de malignidade, a rapidez com que recidivava e a localização totalmente atípica dos casos de angiofibromas.
Por certo a radioterapia representou um fator de indução para o aparecimento deste fenômeno. No exame das lâminas observou-se apenas um aumento na celularidade, porém não surgiram mitoses atípicas, tão próprias das lesões malignas.
No passado vimos ocorrer este mesmo fato nos casos de adenoma pleomorfo, localizado na cavidade oral e que em determinado momento passavam a crescer e a recidivar com grande velocidade. Também o anatomopatológico acompanhado por especialistas experientes nunca mostravam malignidade. A indicação da radioterapia, nestes casos, em geral levam à transformação maligna, como foi o caso por nós observado.
A utilização da radioterapia em pacientes jovens levanta algumas questões quanto às possíveis implicações que podem surgir, como a degeneração maligna, neoplasias malignas induzidas pela radioterapia e o efeito sobre o crescimento do esqueleto craniofacial8. Na situação de extensa invasão cerebral e o envolvimento de estruturas vitais (seio cavernoso, carótida interna, nervo óptico) a radioterapia deve ser considerada como uma opção terapêutica.
CONCLUSÃO
O diagnóstico precoce continua sendo a chave para um bom prognóstico. A falta de diagnóstico ou de recursos técnicos para a solução do caso levou o paciente a aguardar por 14 anos para ser encaminhado para tratamento num centro com maiores recursos. Com a avaliação pela tomografia computadorizada, revelou-se enorme invasão cerebral e o manejamento cirúrgico destes casos é um verdadeiro desafio. Possibilidades de complicações são freqüentes.
A radioterapia não foi efetiva para reduzir o tumor e predispõe o aparecimento de tumor externo na face, altamente recidivante.
REFERÊNCIAS BIBLIOGRÁFICAS
1. ANDREWS, J. C.; FISCH, U.; VALAVANIS, A., AEPPLI, V.; MAKEK, M.S. - The surgical management of extensive nasopharyngeal angiofibroma with infratemporal fossa approach. Laryngoscope, 99:429-37, 1989.
2. ANTONELLI, A.R., CAPPIELLA, J., DONAJO, C.A. et al. - Diagnosis staging and treatment of juvenile nasopharyngeal angiofibroma. Laryngoscope; 97:1319-25, 1987.
3. BATSAKIS, J.G. - Tumours of Head and Neck. Clinical and pathological consideration. (2 ed.) Baltimore: Wiiliams and Wilkis Co., 979:291 312.
4. BHATIA, M.L.; MISHRA, S.C. - Intracranial extensions of juvenile angiofibroma of the nasopharyns. J. Laryngol. Otol., 81:1395-403, 1967.
5. CHANDLER, J.R.; GOLDING, R.; MOSKOWITZ, L.; QUENCER, R.M. -Nasopharyngeal angiofibroma: Staging and management. Ann. Otolaryngol, 93:320-9, 1984.
6. CUMMINGS, B.J.; BLEND, R.; FITZPATRICK, P.; CLARK, R., HARWWOD, A. et al. - Primary radiation therapy for juvenile nasopharyngeal angiofibroma. Laryngoscope, 94:1599-605, 1984.
7. FAGAN, F. F.; SNYDERMAN, C. H.; CARRAU, R. L.; JANECKA, I. P. - Nasopharyngeal Angiofibromas:selecting a surgical approach. Head & Neck 19:391-9, 1997.
8. GRANATO, L. Angiofibroma juvenil - Vias de abordagem, técnicas e complicações. BRANDÃO, L.G.; FERRAZ, A.R. Cirurgia de Cabeça e Pescoço. São Paulo - Livraria Roca. 2:397-414, 1989.
9. GUPTA, A.; MURTHY, D. P. - Intracranial Juvenile Nasopharyngeal Angiofibroma. J. Surg. 7:477-82, 1997.
10. HERMAN, P.; LOT, G.; CHAPOT, R.; SALVAN, D.; TRANBAIAWY, P. - Long-Term follow-up of juvenile nasopharyngeal angiofibromas: Analysis of Recurrences. Laryngoscope 109:140-7, 1999.
11. JACKOBSON, M.; PETRUSON, B.; SVENDENSEN, P., BERTHELSEN, B. - Juvenile nasopharyngeal angiofibroma: A report of 18 cases. Acta Otolaryngol. (Stockh.) 105:132-9, 1988.
12. McGAHAN, R.A.; DURRANCE, F.Y.; PARKE, R.B.; EASLEY, J.D.; CHOU, J.L. - The treatment of advanced juvenile nasopharyngeal angiofibroma. Int. J. Rad. Oncol. Biol. Phys. 17:1067-72, 1989.
13. SPECTOR, J.G. - Management of juvenile angiofibromata. Laryngoscope, 98:1016-26, 1988.
WIATRAK, B.J.; KOOPMANN, C.F.; TURRISI, A.T. - Radiation therapy as na alternative to surgery in the management of intracarnial juvenile nasopharyngeal angiofibroma. Int. J. Pediatr. Otorhinolaryngol., 28(1):51-61, 1993.
1 Prof. Chefe de Clínica do Depto de ORL da Santa Casa de São Paulo.
2 Doutorando do Curso de Pós-Graduação em ORL da Faculdade de Ciências Médicas da Santa Casa de São Paulo.
Trabalho realizado no Depto de ORL da Santa Casa de São Paulo.
Departamento de Otorrinolaringologia da Santa Casa de Misericórdia de São Paulo
Rua Dr. Cesário Mota Junior, 112 - Vila Buarque - SP - 01277-900 - Fone: (11)3226-7235
Artigo recebido em 24 de maio de 2001. Artigo aceito em 29 de junho de 2001.
Imprimir: ![]()