

Ano: 2014 Vol. 80 Ed. 2 - Março - Abril - (4º)
Seção: Artigo Original
Páginas: 99 a 104
 PDF PT
PDF PT Síndrome de Alström familiar: uma rara causa de perda auditiva progressiva bilateral*
Familial Alström syndrome: a rare cause of bilateral progressive hearing loss*
Autor(es): Fayez Bahmad Jr.1,2,3,; Carolina Sousa Alves Costa3; Marina Santos Teixeira2; Jairo de Barros Filho2; Lucas Moura Viana2; Jan Marshall4
DOI: 10.5935/1808-8694.20140023
Palavras-chave: Síndrome de Alström; Perda auditiva neurossensorial; Obesidade infantil; Diabetes mellitus tipo 2; Retinite pigmentosa
Keywords: Alström syndrome; Sensorineural hearing loss; Childhood obesity; Diabetes mellitus, type II; Retinitis pigmentosa
Resumo:
INTRODUÇÃO: A Síndrome de Alstrom é uma doença muito rara, causada pela mutação no gene ALMS1, que apresenta uma degeneração progressiva das funções sensoriais, resultando em deficiências visuais e auditivas, além de distúrbios metabólicos como obesidade na infância, hiperinsulinemia e diabetes tipo II.
OBJETIVO:: Apresentar o perfil audiométrico de dois irmãos da mesma família afetados pela Síndrome de Alström.
MÉTODO: Estudo prospectivo, analítico descritivo, os pacientes afetados foram submetidos a um questionário previamente testado, audiometria tonal e vocal seriadas, análise de emissões otoacústicas, e de respostas de potencial evocado auditivo de tronco encefálico, além de análise genético-molecular para comprovação diagnóstica.
RESULTADOS: Ambos os pacientes apresentaram perda auditiva bilateral com o início na infância e progressão lenta para perda auditiva neurosensorial severa no primeiro caso e, profunda, no segundo. As emissões otoacústicas estavam ausentes, e o potencial evocado auditivo de tronco encefálico estava normal em ambos os pacientes, bilateralmente.
CONCLUSÃO: A Síndrome de Alström apresenta início precoce de perda auditiva neurossensorial, antes da adolescência, 10 a 20 anos para desenvolver perda auditiva severa a profunda. A lesão auditiva é essencialmente coclear, de acordo com os resultados dos testes de emissões otoacústicas e de potenciais evocados auditivos de tronco encefálico.
Abstract:
INTRODUCTION: Alström Syndrome is a rare disease caused by mutations in ALMS1 gene. It is characterized by a progressive degeneration of sensory functions, resulting in visual and audiological impairment, as well as metabolic disturbances such as childhood obesity, hyperinsulinemia, and diabetes mellitus type 2.
OBJECTIVE: To report and discuss the genetic and audiological findings in two siblings with Alström syndrome.
METHODS: This was a prospective, analytical and descriptive study, using questionnaires, serial audiograms, otoacoustic emissions, and auditory brainstem response analysis, as well as molecular genetic analysis.
RESULTS: Both patients presented childhood-onset bilateral sensorineural hearing loss, which progressed to moderate impairment in the first case and severe hearing loss in the second. Otoacoustic emissions were absent, and auditory brainstem responses were bilaterally normal in both cases.
CONCLUSION: In the present patients, Alström Syndrome began with a neurosensory hearing loss in early childhood that progressed to a profound loss in ten to twenty years. The auditory lesions were cochlear in origen according to the otoacoustic emissions and auditory brainstem responses.
![]()
INTRODUÇÃO
De acordo com Alström et al.,1 a Síndrome de Alström é um distúrbio geneticamente transmitido com um leque de sintomas variados, que inclui degeneração sensorial progressiva, resultando em déficits auditivos e visuais, além de obesidade. Outras áreas que apresentam função anormal são: metabólica e endócrina, além de disfunção renal, cardíaca e hepática.
A descrição de casos clínicos na Síndrome de Alström é muito pequena. A primeira publicação de um artigo científico mais valioso foi realizada por Russell-Eggitt et al.,2 que fizeram uma análise longitudinal de 22 casos da síndrome, dando enfoque à histopatologia dos órgãos e outros fatores que levaram ao óbito dos pacientes afetados pela síndrome. Marshall et al.3 apresentaram 182 casos afetados pela síndrome. Segundo Welsh et al.,4 o fator genético que causa a síndrome foi identificado como uma translocação recíproca em um novo gene de desconhecida função, ALMS1, localizado no cromossomo 2p13.
Mesmo que a Síndrome de Alström tenha sido descrita como uma alteração homogênea decorrente da mutação de um único gene, existem famílias descritas com apresentações clínicas variáveis. Ocorre desde cardiomiopatia, disfunção hepática, hipotireoidismo e hipogonadismo. Esses sintomas variam em indivíduos de uma mesma família, devido a interações dos modificadores genéticos.1 Collin et al.5,6 comentaram em seu artigo que o fenótipo da Síndrome de Alström seria decorrente da função alterada, mais do que o desenvolvimento inadequado dos órgãos.
Alström et al.1 relataram o início da deficiência auditiva até os dez anos de idade em pacientes afetados pela síndrome, e que seria progressiva desde então. Michaud et al.7 confirmaram que a surdez iniciava-se na primeira década de vida. Marshall et al.3 indicaram que a deficiência auditiva surgia durante a infância, e a média de aparecimento dos sintomas era de cinco anos de idade. Coincidentemente, otite média crônica serosa foi encontrada em 42% de seus pacientes, adicionando um componente condutivo à já descrita lesão neurossensorial.7 Russell-Eggitt et al.2 relataram que somente dois de seus 22 casos apresentavam audição normal após 12 anos de idade.
O objetivo deste estudo foi realizar ampla revisão bibliográfica e apresentar, de forma inédita, o perfil clínico audiométrico, além de análise genética molecular, dos dois únicos casos relatados até esta data na América do Sul de irmãos da mesma família afetados pela Síndrome de Alström, de forma que isso propicie um conhecimento maior desta doença tão rara.
MATERIAIS E MÉTODOS
Foi realizado um estudo prospectivo, analítico descritivo do fenótipo de uma família caucasiana, no Brasil, com membros afetados pela Síndrome de Alström. Os dados foram obtidos através de uma avaliação em longo prazo dos pacientes no consultório, revisão de prontuários e um questionário respondido pelos familiares.
Os pacientes foram submetidos a uma avaliação otorrinolaringológica criteriosa, com análise audiológica por meio de audiometria tonal e vocal, imitanciometria, emissões otoacústicas por produto de distorção (EOAPD) e transiente (EOAT), e potencial evocado auditivo de tronco encefálico (PEATE).
Foi realizada audiometria tonal liminar por via aérea, nas frequências de 250 a 8000 Hz e por via óssea, nas frequências de 500 a 4000 Hz; índice de reconhecimento de fala e limiar de reconhecimento de fala, utilizando o equipamento GSI68, equipado com fones TDH-39. Foi realizada a timpanometria, utilizando o equipamento GSI38. Para esta avaliação foram empregados os critérios propostos por Russo e Santos.8
EOAPD e EOAT foram realizadas no aparelho da marca Biologic, modelo Scout. Foram avaliadas as amplitudes e a relação sinal/ruído de ambas as orelhas, baseados nos critérios de Figueiredo9 e Gorga.10 PEATE foi realizado no equipamento da marca Biologic, modelo Navigator-Pro. Os eletrodos foram colocados de acordo com o sistema internacional de eletrodos:11 o ativo no vértex (Cz), o terra, na fronte (Fz) e os de referência nos lóbulos das orelhas direita e esquerda (A1 e A2). O fone utilizado foi o TDH-39. O tipo de estímulo foi o clique com intensidade de 80 dB NA. Foram pesquisadas as latências absolutas das ondas I, III e V, as latências interpicos das ondas I-III, III-V e I-V, e a comparação binaural da latência da onda V. Para esta avaliação foram empregados os critérios de Hood.12
Para a realização do diagnóstico genético-molecular, o termo de consentimento foi obtido de todos os pacientes envolvidos na pesquisa, incluindo seus familiares. Este estudo foi aprovado pelo comitê de ética em pesquisa da instituição onde foi realizado com o protocolo número 1112000005. DNA genômico, isolado de sangue periférico em acordo com o método padrão, foi amplificado usando o protocolo de Reação de polimerase em cadeia (PCR), como previamente descrito.10 Sequências de primers e as condições de amplificação estão disponíveis caso solicitadas através do contato do autor. PCR amplicons dos exons 8, 10 e 16 do gene ALMS1 foi purificado, diretamente sequenciado e analisado por um sistema chamado ABI 3730 Sequencing Analyzer (Applied Biosystems, Forster City, CA, USA). As sequências resultantes foram comparadas com as de referência do GenBank mRNA (NM_015120.4). A nomenclatura das mutações foi definida por den Dunnen e Antonarakis.11
RESULTADOS
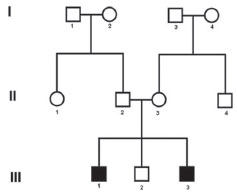
Os dois pacientes afetados apresentam surdez neurossensorial progressiva, cujos sintomas se iniciaram por volta dos seis anos de idade. A figura 1 revela o heredograma da família. Tomografia computadorizada de mastoides e ressonância nuclear magnética foram realizadas em ambos os pacientes, mas não apresentaram alterações. Foram negativas para aqueduto vestibular alargado ou algum grau de displasia cocleosacular.
Figura 1 Pedigree mostra os dois afetados pela Síndrome de Alström (caso 1: III-1 e caso 2: III-3).
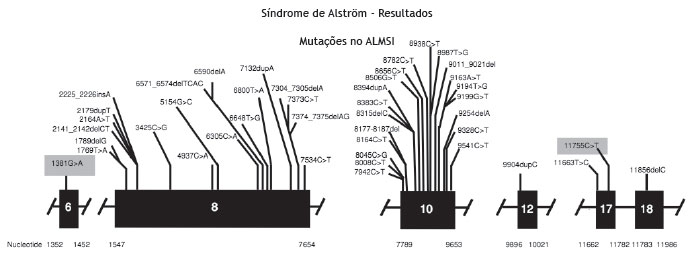
A análise genética confirmou a mutação no gene ALMS1 nos dois irmãos. Duas mutações heterozigóticas foram identificadas no exon 10 ALMS1 (c.7942C > T, Gln2648* e c.9163ª > T, Lys3055*), confirmando o diagnóstico de Síndrome de Alström (fig. 2).
Figura 2 Mutação evidenciada no ALMS1 gene.
O paciente 1 tinha 30 anos, portador de fotofobia desde o nascimento, nistagmo com aparecimento aos seis meses de idade e retinose pigmentar desde os dez. Não apresentou otite média de repetição. É portador de cardiomiopatia dilatada, diagnosticada aos 28 anos de idade, porém, com ausência de insuficiência cardíaca congestiva. Não apresenta sintomas pulmonares, hepáticos e renais. Apresenta diabetes com resistência à insulina desde os 27 anos de idade, além de hiperinsulinemia, hipertrigliceridemia, hipogonadismo, hipotireoidismo, acanthosis nigricans e obesidade desde a infância.
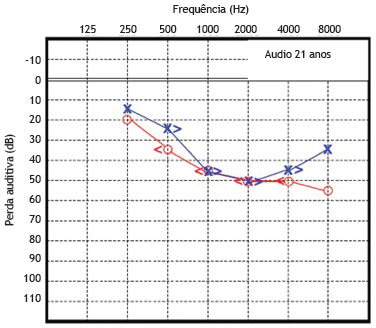
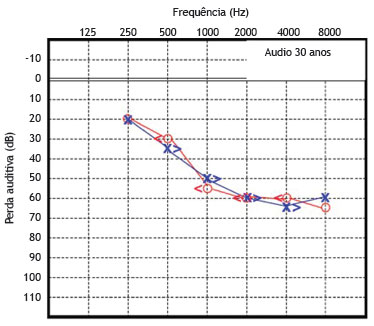
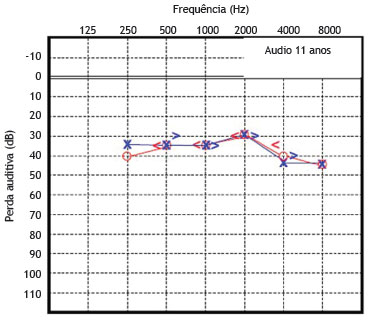
O paciente 1 foi examinado pela primeira vez e diagnosticado como portador de surdez neurossensorial de causa genética não esclarecida. A função coclear na idade de seis anos foi testada, revelando uma surdez neurossensorial moderada bilateralmente. A partir de então, este paciente foi submetido a avaliações seriadas, com audiometrias tonais e vocais. As figuras 3 e 4 mostram os resultados das audiometrias tonais aos 21 e 30 anos de idade, respectivamente.
Figura 3 Audiometria do paciente 1 realizada aos 21 anos de idade.
Figura 4 Audiometria do paciente 1 realizada aos 30 anos de idade.
Essas avaliações audiométricas seriadas revelaram uma perda auditiva neurossensorial de progressão lenta, bilateralmente. Aos 28 anos, o déficit auditivo requereu o uso de aparelho de amplificação sonora individual bilateralmente, apresentando melhora parcial da acuidade auditiva em ambas as orelhas.
Avaliação audiométrica recente, feita aos 30 anos de idade, revela perda média de 50 dB NA para as orelhas direita e esquerda, conforme demonstrado na figura 3. A timpanometria revelou curvas normais, tipo "A", para ambas as orelhas.
Os resultados recentes de emissões otoacústicas, para o paciente 1, OEAPD e OEAT revelaram-se ausentes em ambas as orelhas. Foram analisadas as amplitudes e a relação sinal/ruído bilateral, e estas apresentam-se abaixo do esperado quando comparadas com indivíduos normais, mostrando função coclear alterada.
Foi realizado, também, o PEATE, em intensidade de 80 dBNA, que revelou-se normal para as duas orelhas, com presença das ondas I, III e V. As latências absolutas das ondas I, III e V, e latências interpicos das ondas I-III, III-V e I-V estão dentro dos padrões de normalidade, assim como a comparação binaural da latência da onda V.
O paciente 2 tinha 26 anos e apresenta fotofobia desde o nascimento, nistagmo ainda com seis meses de vida, e retinose pigmentar diagnosticada ainda criança. Apresentou otites de repetição desde o início da infância, além de otalgia e otorreia. Apresentou uma cardiomiopatia dilatada aos dois meses de idade, com ausência de insuficiência cardíaca congestiva. Fez controle durante um ano e esta desapareceu completamente. Não apresenta sintomas pulmonares e renais. É portador de uma disfunção hepática diagnosticada aos 20 anos de idade. Não apresenta diabetes, porém é portador de hiperinsulinemia, hipertrigliceridemia, hipogonadismo, hipotireoidismo e obesidade desde a infância. Não apresenta achantosis nigricans.
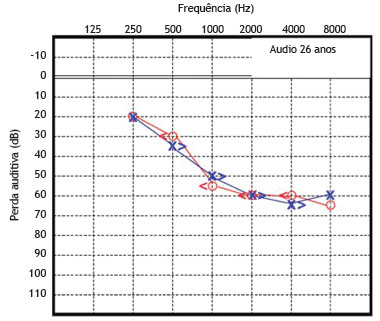
O paciente 2 foi examinado pela primeira vez e diagnosticado como portador de surdez neurossensorial de causa genética não esclarecida, como seu irmão mais velho. A função coclear na idade de seis anos foi testada e apresentou-se alterada. A partir de então, este paciente foi submetido a avaliações seriadas, além de audiometrias tonais e vocais (figs. 5 e 6).
Figura 5 Audiometria do paciente 2 realizada aos 11 anos de idade.
Figura 6 Audiometria do paciente 2 realizada aos 26 anos de idade.
Essas avaliações audiométricas seriadas revelaram uma perda auditiva neurossensorial de progressão lenta, bilateralmente. Com a idade de 23 anos, o déficit auditivo requereu o uso de aparelho de amplificação sonora individual bilateralmente, apresentando melhora parcial da acuidade auditiva em ambas as orelhas. A avaliação audiométrica recente, feita aos 26 anos de idade, revela perda média de 70 dB NA para a orelha direita e 65 dB NA para a esquerda. A timpanometria apresentou curvas do tipo "As" bilateralmente.
EOAPD e EOAT revelaram-se ausentes em ambas as orelhas. Foram analisadas as amplitudes e a relação sinal/ruído bilateral, e estas se apresentaram abaixo do esperado, quando comparadas com indivíduos normais, mostrando função coclear alterada.
O exame de potencial evocado auditivo de tronco encefálico, em intensidade de 80 dB NA, revelou-se normal para as duas orelhas, com presença das ondas I, III e V. As latências absolutas das ondas I, III e V e latências interpicos das ondas I-III, III-V e I-V encontravam-se dentro dos padrões de normalidade, assim como a comparação binaural da latência da onda V.
DISCUSSÃO
Os relatos limitados concernentes à Síndrome de Alström e os poucos casos apresentados na literatura mundial sugerem a raridade desta condição. O início precoce da surdez neurossensorial, que chega à profunda ainda na terceira década de vida, é, geralmente, o sinal mais marcante desta síndrome.
Neste estudo, foram apresentados dois membros de uma mesma família caucasiana que apresentaram um quadro clínico de perda auditiva neurossensorial bilateral, com progressão lenta, iniciando-se aos seis anos de idade. Avaliação audiométrica realizada aos 30 anos de idade, no paciente 1, e aos 26 anos de idade, no caso 2, mostrou surdez neurossensorial moderada e severa, respectivamente. EOAPD e EOAT estavam ausentes e a relação sinal/ruído foi abaixo do normal, quando comparados com indivíduos normais, em ambos os casos, ao passo que a timpanometria e o PEATE foram normais. Esses dados sugerem que a lesão causadora da perda auditiva é coclear.
A etiologia da surdez neurossensorial na infância tem varias causas, incluindo causas hereditárias, infecciosas, relacionadas ao desenvolvimento, além de outras menos comuns, tais como autoimune, traumática e vascular.
As causas hereditárias podem ser divididas em não sindrômicas e sindrômicas, sendo as últimas menos prevalentes.13 Entre as causas sindrômicas, podemos citar a Síndrome de Usher como sendo o mais importante diagnóstico diferencial de perda auditiva com início precoce e lenta progressão, associado à perda visual, porém quais seriam as melhores indicações sugestivas de Síndrome de Alström em pacientes portadores de perda auditiva neurossensorial progressiva de início precoce?
Primeiro, por meio das avaliações audiométricas seriadas (sensibilidade tonal), que podem confirmar o caráter lentamente progressivo desta síndrome, que se desenvolve por mais de 15 anos, levando à perda auditiva profunda. Sinais de retinopatia, facilmente identificáveis por meio da retinoscopia e confirmadas pela eletroretinografia, constituem outro ponto que facilita o diagnóstico da síndrome.
Além destes aspectos audiológicos e visuais, a análise de distúrbios metabólicos, tais como diabetes mellitus tipo 2, hipertrigliceridemia, hipogonadismo, hipotireoidismo, disfunção renal e hepática e obesidade desde a infância, apesar de serem variáveis entre os pacientes afetados, também podem servir para o diagnóstico diferencial entre outras síndromes.
Uma vez que a Síndrome de Alström é causada pelas mutações no gene ALMS1, a análise genético-molecular deve ser empregada para confirmar o diagnóstico clínico.14-17
Esta é a primeira descrição em língua portuguesa de dois membros de uma mesma família afetados pela síndrome de Alström. Enquanto ainda não existem publicações científicas revelando a histopatologia da cóclea nesta síndrome, sugerimos que, por detrás desta degeneração lentamente progressiva da audição, existe um mecanismo que, como resultado final, leve à destruição das células ciliadas no órgão de Corti.
Embora não haja tratamento preventivo para a surdez neurossensorial em pacientes com Síndrome de Alstrom, a sua identificação é importante para que possa ser realizado o aconselhamento familiar. Alem do mais, o seguimento desses pacientes com uma equipe multidisciplinar pode ajudá-los a adquirir um melhor desenvolvimento educacional e psicossocial, além da reabilitação e do tratamento das desordens associadas. A identificação precoce da perda auditiva e a sua pronta reabilitação através do uso de aparelhos de amplificação sonora individual bilateral podem permitir aos pacientes um melhor desenvolvimento da linguagem, tornando-os bons candidatos para reabilitação auditiva com o uso da tecnologia do implante coclear.
CONCLUSÃO
Após longo período de seguimento (mais de 20 anos), observamos que o perfil audiométrico desta síndrome pode ser caracterizado pelo início precoce da perda auditiva neurossensorial, geralmente na primeira década de vida, progressão lenta, levando de 10 a 20 anos para provocar perda auditiva profunda nos afetados.
O provável local da lesão é coclear, fato sugerido pelas análises dos testes de emissões otoacústicas e do potencial evocado auditivo de tronco encefálico.
Apesar da raridade da síndrome, a sua identificação precoce e o tratamento da perda auditiva, através do uso de aparelhos amplificação sonora individual bilateral, pode permitir aos pacientes um adequado desenvolvimento da linguagem.
FINANCIAMENTO
Este estudo foi conduzido pelo CNPQ - Lucas Moura Viana - Bolsa de Doutorado - Programa de Pós-graduação em Ciências da Saúde - Universidade de Brasília.
CONFLITOS DE INTERESSE
Os autores declaram não haver conflitos de interesse.
REFERÊNCIAS
1. Alstrom CH, Hallgren B, Nilsson LB, Asander H. Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the laurence-moon-bardet-biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree. Acta Psychiatr Neurol Scand Suppl. 1959;129:1-35.
2. Russell-Eggitt IM, Clayton PT, Coffey R, Kriss A, Taylor DS, Taylor JF. Alstrom syndrome. Report of 22 cases and literature review. Ophthalmology. 1998;105:1274-80.
3. Marshall JD, Bronson RT, Collin GB, Nordstrom AD, Maffei P, Paisey RB, et al. New Alstrom syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med. 2005;165:675-83.
4. Welsh LW. Alstrom syndrome: progressive deafness and blindness. Ann Otol Rhinol Laryngol. 2007;116:281-5.
5. Collin GB, Cyr E, Bronson R, Marshall JD, Gifford EJ, Hicks W, et al. Alms1-disrupted mice recapitulate human Alstrom syndrome. Hum Mol Genet. 2005;14:2323-33.
6. Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nat Genet. 2002;31:74-8.
7. Michaud JL, Heon E, Guilbert F, Weill J, Puech B, Benson L, et al. Natural history of Alstrom syndrome in early childhood: onset with dilated cardiomyopathy. J Pediatr. 1996;128:225-9.
8. Russo ICP, Santos TMM. A Prática da Audiologia Clínica. 4º ed. São Paulo: Cortez Editora; 1993.
9. Figueiredo MS. Emissões Otoacústicas e Bera (Coleção CEFAC). São Paulo: Pulso Editora; 2003.
10. Gorga MP, Stover L, Neely ST, Montoya D. The use of cumulative distribuitions to determine critical values and levels of confiance for clinical distortion product otoacoustic emission. J Acoust Soc Am. 1996;100:968-77.
11. Klem GH, Lüders HO, Jasper HH, Elger C. The ten-twenty electrode system of the International Federation. The International Federation of Clinical Neurophysiology. Electroencephalogr Clin Neurophysiol Suppl. 1999;52:3-6.
12. Hood LJ. Clinical Applications of the auditory brainstem response. San Diego, CA: Singular Publishing Group, Inc; 1998.
13. Ruben RJ. Diseases of the inner ear and sensorineural deafness. In: Bluestone CD, Stool SE, Scheetz MD, eds. Pediatric otolaryngology, Vol. 1. Philadelphia: WB Saunders; 1990. p. 547-70.
14. Tseng CJ, Lalwani AK. Cracking the auditory genetic code: part IL Syndromic hereditary hearing impairment. Am J Otol. 2000:21:437-51.
15. Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat Genet. 2002;31:74-8.
16. Hearn T, Renforth GL, Spalluto C, Hanley NA, Piper K, Brickwood S, et al. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alström syndrome. Nat Genet. 2002;31:79-83.
17. Marshall JD, Hinman EG, Collin GB, Beck S, Cerqueira R, Maffei P, et al. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alström syndrome. Hum Mutat. 2007;28:1114-23.
1. Faculdade de Medicina, Universidade de Brasília (UnB), Brasília, DF, Brasil
2. Programa de Pós-graduação, Faculdade de Ciências da Saúde, Universidade de Brasília (UnB), Brasília, DF, Brasil
3. Instituto Brasiliense de Otorrinolaringologia, Brasília, DF, Brasil
4. The Jackson Laboratory, Maine, USA
Autor para correspondência.
F. Bahmad Jr
E-mail: fayez@unb.br
Recebido em 4 de novembro de 2012.
Aceito em 30 de agosto de 2013.
* Instituição: Faculdade de Ciências da Saúde da Universidade de Brasília, Brasília, DF, Brasil.
Imprimir: ![]()