

Ano: 2002 Vol. 68 Ed. 4 - Julho - Agosto - (20º)
Seção: Relato de Caso
Páginas: 581 a 585
 PDF PT
PDF PT Agenesia do nariz: Relato de Caso
Agenesis of the nose: Case Report
Autor(es):
Daniel Chung 1,
Elder Y. Goto 2,
Fabrízio R. Romano 2,
Marcus M. Lessa 2,
Richard L. Voegels 3,
Ossamu Butugan 4
Palavras-chave: agenesia do nariz, arrinia, tomografia computadorizada e ressonância nuclear magnética
Keywords: agenesis of the nose, arhinia, computed tomography, magnetic resonance imaging
Resumo:
A agenesia do nariz (arrinia, atresia nasal congênita) é uma malformação rara, tendo sido relatados aproximadamente 20 casos na literatura de língua inglesa no último século. Acredita-se que a origem embriológica do defeito seja o subdesenvolvimento dos placódios nasais. As dificuldades respiratórias se evidenciam logo após o nascimento e requerem intubação orotraqueal imediata seguida de ventilação mecânica. Neste trabalho relatamos um caso de ausência congênita do nariz em uma criança do sexo masculino. Logo após o nascimento, apresentou desconforto respiratório, evoluindo rapidamente com cianose, sendo submetida a intubação orotraqueal e posteriormente traqueostomia. Ao exame físico apresentava ausência completa da pirâmide nasal, substituída por uma superfície plana e lisa de consistência endurecida, palato duro em forma de ogiva e microftalmia bilateral. A criança foi avaliada também através de exames de tomografia computadorizada (TC) e ressonância nuclear magnética (RNM). A TC revelou ausência do nariz, estenose óssea anterior, ausência de cavidade nasal e seios paranasais, palato arqueado em ogiva, microftalmia bilateral e mucocele bilateral de saco lacrimal. A RNM demonstrou conFiguração e densidade normal do tecido cerebral.
Abstract:
Agenesis of the nose (arhinia, congenital nasal atresia) is a rare anomaly with about 20 surviving cases reported in the English literature in the last century. The embryological origin of the defect is thought to be maldevelopment of the paired nasal placodes. The respiratory distress is evident after the birth and requires immediate orotracheal intubation and mechanical ventilation. Herein we report a case of congenital absence of the nose in a male child. Immediately after birth, the baby had difficulty in breathing and developed cyanosis, for which he required orotracheal intubation and tracheotomy, posteriorly. On examination a, a flat bony area replaced the nose, the hard palate was highly arched, and bilateral microphthalmia. The child was also was evaluated by CT (computed tomography) and MRI (magnetic resonance imaging) studies. The CT scan revealed absence of the nose, anterior bony stenosis, absence of the nasal chamber and paranasal sinuses, a high-arched palate, bilateral microphtalmia, and bilateral mucocele of the lachrymal sac. The MRI demonstrated normal conFiguration and density of the brain.
![]()
INTRODUÇÃO
A ausência congênita do nariz, também conhecida pelo termo arrinia ou atresia congênita nasal, é uma malformação muito rara e pouco descrita na literatura médica2. Geralmente está associada a outras malformações, principalmente no sistema nervoso central, e apresenta alta taxa de mortalidade7.
O primeiro caso foi descrito em 1903 por Wahby12, que notou a ausência dos ossos nasais e da pré-maxila ao examinar um crânio da coleção do Museu da Universidade de Cambridge. Desde então, foram publicados cerca de vinte casos da referida malformação na literatura médica de língua inglesa7.
De acordo com Rosen10, a ausência congênita do nariz pode ser classificada em arrinia e arrinia total, sendo a primeira caracterizada pela ausência da pirâmide nasal e a última pela ausência do nariz associada à agenesia do sistema olfatório.
Cada caso de arrinia merece ser cuidadosamente estudado, pois, por se tratar de uma malformação muito rara, ainda não apresenta etiologia e tratamento claramente definidos.
RELATO DE CASO
Este relato refere-se a uma criança da raça parda, do sexo masculino e com a idade de 2 anos. Essa criança havia sido atendida em outro Serviço e nos foi encaminhada para uma avaliação mais minuciosa de sua malformação de face.
A genitora da criança, de 35 anos de idade, era mãe de outras 6 crianças sem quaisquer malformações e outras doenças de relevância. Apesar de não ter feito um acompanhamento pré-natal adequado, negava intercorrências durante a gestação de 40 semanas. Nega, ainda, consangüinidade com o pai da criança, doenças anteriores à gestação (diabetes, hipertensão, endocrinopatias), etilismo, tabagismo, abortos prévios e casos de malformação na família.
O parto ocorreu em 16 de fevereiro de 1997, sendo espontâneo por via vaginal, tendo o recém-nascido chorado logo após o parto, evoluindo, porém, com cianose e dificuldade respiratória, sendo imediatamente submetida a intubação orotraqueal. Ao exame físico apresentava ausência completa da parte externa do nariz e microftalmia bilateral. Durante o primeiro mês de vida permaneceu sob cuidados intensivos, sendo precocemente traqueostomizada com o intuito de prevenir complicações respiratórias, já que o Hospital não dispunha de recursos para a cirurgia reparadora definitiva.
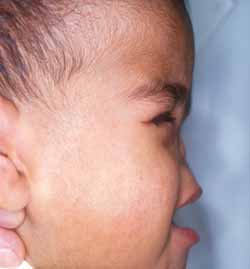
Em abril de 1999, a criança de 2 anos de idade foi recebida em nosso Serviço, onde verificamos (Figura 1): ausência completa da pirâmide nasal, substituída por uma superfície plana e lisa de consistência endurecida, palato duro alto e ogival, lábios e filtro labial sem alterações morfológicas, otoscopia normal, microftalmia bilateral e ausência de déficit motor ou cognitivo. Apresentava amaurose, porém sua avaliação audiométrica, alimentação e comunicação eram normais.
Durante seu acompanhamento realizamos a retirada da cânula de traqueostomia 2 meses após sua chegada ao nosso Serviço, tendo o traqueostoma cicatrizado normalmente. Realizamos a investigação radiológica, através da tomografia computadorizada (TC) dos seios paranasais e ressonância nuclear magnética (RNM).
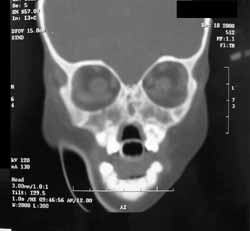
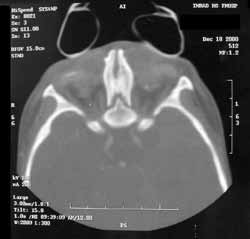
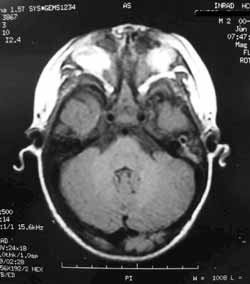
A TC dos seios paranasais (Figuras 2 e 3) evidenciou agenesia de pirâmide nasal; cavidade nasal residual, observada como um pequeno espaço na porção superior do osso maxilar preenchido por material com densidade de partes moles; palato duro em ogiva, com pequena fenda; e presença de palato mole delimitando um diminuto espaço nasofaríngeo posteriormente. Constatou-se, também, ausência de ductos naso-lacrimais, de seios paranasais e de falhas ósseas em base de crânio. Os globos oculares apresentavam dimensões acentuadamente reduzidas com deformidades em suas paredes posteriores e calcificações à esquerda. Na topografia dos sacos lacrimais foram encontradas duas formações císticas de aproximadamente 1,5 cm., sugerindo o diagnóstico de mucocele.
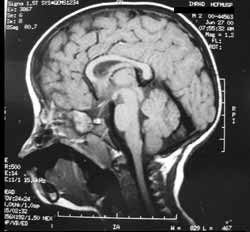
A RNM de face e crânio (Figuras 4, 5 e 6) demonstrou, além das deformidades supracitadas, rebaixamento dos giros frontais basais na topografia das fóveas etmoidais, com demais estruturas encefálicas sem alterações.
No momento, a criança aguarda uma abordagem cirúrgica conjunta entre a equipe de Otorrinolaringologia e de Cirurgia Plástica, com o objetivo de reconstruir o nariz externo e criar uma comunicação aérea até a rinofaringe.
Figura 1. Fotografia da criança em perfil.
Figura 2. Tomografia computadorizada de seios paranasais: corte coronal em janela óssea.
Figura 3. Tomografia computadorizada de seios paranasais: corte axial em janela óssea.
Figura 4. Ressonância nuclear magnética de face: corte sagital em T1.
Figura 5. Ressonância nuclear magnética de face: corte coronal em T2.
Figura 6. Ressonância nuclear magnética de face: corte axial em T1.
EMBRIOLOGIA
Os primórdios da face começam a surgir no início da 4ª semana do desenvolvimento embrionário em torno do estomodeu (boca primitiva), que se localiza no 1º arco branquial. A proeminência frontonasal (PFN), situada logo acima do estomodeu, dá origem ao nariz e à região frontal da face. No fim da 4ª semana, formam-se espessamentos bilaterais do ectoderma ventro-lateral da PFN (placódios nasais) e através da proliferação do mesênquima na margem desses placódios, forma-se uma estrutura em forma de ferradura, que pode ser didaticamente dividida em proeminências nasais medial e lateral. No centro do placódio notamos uma depressão conhecida como fosseta nasal, que representa o primórdio da narina e da cavidade nasal. O ducto naso-lacrimal desenvolve-se a partir de um espessamento do ectoderma (em forma de bastão) no assoalho do sulco naso-lacrimal, que se aprofunda no mesênquima unindo o saco lacrimal à cavidade nasal. Entre a 7ª e 10ª semana ocorre a fusão das proeminências nasais mediais entre si, formando o filtro do lábio superior, e a formação das coanas primitivas através das fossetas nasais, que se aprofundam e rompem a membrana oronasal8.
De acordo com Meyer7, a arrinia decorre de uma falha no desenvolvimento dos placódios nasais. Os ductos naso-lacrimais se formam inicialmente, mas terminam em fundo cego, já que não conseguem se comunicar à cavidade nasal. O palato assume um aspecto arqueado e alto não só pela falta da cavidade nasal, mas também pela ausência do crescimento do septo nasal, que contribui para a formação normal do palato secundário. O epitélio olfatório, como poderia se prever, não se forma na ausência da invaginação da fosseta nasal, levando à hipoplasia dos bulbos olfatórios e anosmia.
As deformidades oculares associadas são raras e provavelmente causadas pelo subdesenvolvimento do aparelho naso-lacrimal13. A etiologia exata dessa malformação associada não é clara e talvez seja mais complexa, já que existem casos relatados na literatura com arrinia isoladamente2,4,7,11.
DISCUSSÃO
Apesar de existirem algumas teorias, baseadas na embriologia, que explicam a ausência congênita do nariz, nenhuma causa etiológica convincente foi aventada até o presente momento. De acordo com um levantamento de 12 casos de arrinia, Cohen3 notou que os principais fatores associados foram: diabetes materno (2 casos), polidrâmnio (2 casos), anomalias no cromossomo 9 (2 casos) e história de sangramento materno no início da gestação (2 casos). Quanto ao nosso paciente, não se pode afirmar se apresenta anomalia no cromossomo 9, já que não foi realizado o estudo do cariótipo; porém, é certo que não apresenta nenhum desses outros fatores, assim como o caso relatado por Cole4.
É importante observar que seguindo a classificação sugerida por Rosen10, podemos definir este caso como uma arrinia total, já que não há sinais de formação dos nervos e bulbos olfatórios.
Os exames de imagem são de importância fundamental para o estudo da ausência congênita do nariz. O diagnóstico pode ser feito mesmo antes do parto, como demonstrado por Cusick5 em exames ultrassonográficos seriados que indicam o defeito ao se estudar o perfil do feto. A TC pode nos mostrar as alterações ósseas e delimitar margens antes do ato cirúrgico, enquanto que a RNM auxilia na diferenciação entre tecidos de partes moles, que eventualmente podem ser confundidos com encefalocele. A primeira avaliação utilizando esses dois exames ocorreu em 19894 e desde então todos os casos têm sido estudados dessa maneira.
Algumas alterações anatômicas vistas em nosso paciente já haviam sido descritas anteriormente, principalmente estenose óssea anterior1,4,7,9, cavidade nasal hipoplásica4, palato em ogiva4,7,9 e com fenda13, agenesia de ducto naso-lacrimal6,9, hipoplasia de seios paranasais4,9 e microftalmia13. Outras características presentes na literatura médica, que no entanto, não se encontravam em nosso caso são: fenda óssea na placa cribiforme e plano esfenoidal4 e presença de seios maxilares7. Os achados inéditos em nosso paciente consistiram nas duas formações císticas localizadas na topografia dos sacos lacrimais, que suspeitamos serem mucoceles, e nas calcificações encontradas no interior do globo ocular esquerdo.
A RNM da face e do crânio, em geral, demonstra parâmetros normais no sistema nervoso central, com exceção do aparelho olfatório, que na maioria das vezes é hipoplásico1.
Em relação ao tratamento, desde que a primeira tentativa de correção cirúrgica foi realizada2, a grande maioria dos autores têm optado pelo tratamento cirúrgico reparador estético e funcional ou apenas para obter uma passagem aérea fisiológica (neo-cavidade)4. Muitos autores preferem abordar o problema anatômico diretamente, ao invés de realizar a traqueostomia. Essa primeira abordagem em geral é realizada no primeiro mês de vida, com relatos variando entre 15 dias4 e 2 meses13.
A técnica cirúrgica ainda não está plenamente estabelecida e varia bastante, conforme podemos perceber nos relatos de Meyer7 e Mühlbauer9. O primeiro autor realizou a reconstrução do nariz em três tempos cirúrgicos, sendo que o primeiro deles consistiu na criação de um nariz externo, através de um flap cutâneo de região frontal com base inferior, posicionado sobre a região da agenesia nasal, de maneira a recobrir um enxerto de cartilagem costal, que sustentava a estrutura como um neo-septo. A segunda intervenção ocorreu 45 dias após e se resumiu à abertura de duas passagens ósseas, deixando um septo entre eles, exceto na região das coanas, que foram comunicadas. A última cirurgia foi realizada apenas com o intuito de ampliar as fossas nasais. Mühlbauer9, por sua vez, primeiramente implantou 2 expansores sob a pele da região da agenesia. Após 2 meses, realizou a abertura das fossas nasais e a reconstrução da pirâmide nasal em um só tempo cirúrgico. A sua técnica se iniciou com o broqueamento ósseo, formando duas cavidades. Logo após, foram realizadas incisões sobre a pele expandida, de forma a desenhar as narinas e a columela, e um flap de osso frontal mais periósteo foi deslocado para a região do defeito, com o objetivo de manter a arquitetura do nariz externo.
A tendência atual é de se tratar cirurgicamente todos os casos antes que as crianças ingressem na escola, pois, apesar de conseguirem se alimentar e respirar pela via oral6, devemos tentar minimizar os futuros traumas psicológicos que certamente podem surgir.
CONSIDERAÇÕES FINAIS
A arrinia é uma malformação extremamente rara, que geralmente cursa com dificuldade respiratória importante, necessitando de uma intervenção rápida de suporte de vida (intubação orotraqueal ou traqueostomia). A investigação radiológica auxilia na determinação exata das alterações anatômicas e consiste na TC dos seios paranasais e na RNM da face e do crânio. Este defeito do desenvolvimento pode colocar em risco a vida da criança, porém a abordagem cirúrgica definitiva pode ser eletiva, já que a criança se adapta à respiração oral.
REFERÊNCIAS BIBLIOGRÁFICAS
1. ALBERNAZ, V.S.; CASTILLO, M.; MUKHERJI, S.K.; IHMEIDAN, I.H. - Congenital arhinia. AJNR Am. J. Neuroradiol.; 17(7): 1312-4, 1996 Aug.
2. COHEN, D.; GOITEIN, K. - Arhinia. Rhinology; 24: 287-292, 1986.
3. COHEN, D.; GOITEIN, K. - Arhinia revisited. Rhinology, 25: 237, 1987.
4. COLE, R.R.; MYER III, C.M.; BRATCHER, G.O. - Congenital absence of nose: a case report. Int. J. Pediatric Otolaryngol.; 17: 171-178, 1989.
5. CUSICK, W.; SULLIVAN, C.A.; ROJAS, B.; POOLE, A.E.; POOLE, D.A. - Prenatal diagnosis of total arhinia. Ultrasound Obstet. Gynecol.; 15(3): 259-61, 2000 Mar.
6. GALETTI, R.; DALLARI, S.; BRUZZI, M.; VINCENZI, A.; GALETTI, G. - Considerazioni di fisiopatologia respiratoria a proposito di un casi di arinia. Acta Otorhinol. Ital.; 14: 63-69, 1994.
7. MEYER, R. - Total external and internal construction in arhinia. Plast. Reconstr Surg.; 99 (2): 534-542, 1997.
8. MOORE, K.L.; PERSAUD, T.V.N. - Embriologia clínica, Quinta edição, Rio de Janeiro, Editora Guanabara Koogan S. A., 1994, 177-213.
9. MÜHLBAUER, W.; SCHMIDT, A.; FAIRLEY, J. - Simultaneous construction of an internal and external nose in na infant with arhinia. Plast. Reconstr. Surg.; 91 (4): 720-725, 1993.
10. ROSEN, Z. - Embryological introduction to congenital malformations of the nose. Int. Rhinol.; 1: 10, 1963.
11. SMITH, D.W.; DICKSHEET, S. - Reconstruction of agenesis of the external nose secondary to congenital hypertelorism. Clinics Plastic. Surg.; 8 (3): 615-618, 1981.
12. WAHBY, B. - Congenital absence of the nose and premaxilla. J. Anat. 38: 49, 1903.
13. WEINBERG, A.; NEUMAN, A.; BENMEIR, P.; LUSTHAUS, S.; WEXLER, M.R. - A rare case of arhinia with severe airway obstruction: case report and review of the literature. Plast. Reconstr. Surg.; 91 (1): 146-149, 1993.
1 - Médico Residente da Disciplina de Clínica Otorrinolaringológica do Hospital das Clínicas
da Faculdade de Medicina da Universidade de São Paulo.
2 - Médicos Pós-Graduandos da Disciplina de Clínica Otorrinolaringológica do Hospital das
Clínicas da Faculdade de Medicina da Universidade de São Paulo.
3 - Professor Doutor da Disciplina de Clínica Otorrinolaringológica do Hospital das
Clínicas da Faculdade de Medicina da Universidade de São Paulo.
4 - Professor Associado da Disciplina de Otorrinolaringologia da Faculdade de Medicina da
Universidade de São Paulo.
Trabalho realizado na Divisão de Clínica Otorrinolaringológica do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo.
Endereço para correspondência: Daniel Chung
Av. Dr. Enéas de Carvalho Aguiar, 255 - 6°andar - sala 6021
05403-000 - São Paulo - SP - Tel: (0xx11) 3069-6288 Fax: (0xx11) 270-0299.

Imprimir: ![]()