

Ano: 2013 Vol. 79 Ed. 1 - Janeiro - Fevereiro - (16º)
Seção: Artigo Original
Páginas: 95 a 99
 PDF PT
PDF PT Estudo de mutações no gene GJB2 e deleção delGJB6-D13S1830 em indivíduos com surdez não sindrômica da região Amazônica
A study of GJB2 and delGJB6-D13S1830 mutations in Brazilian non-syndromic deaf children from the Amazon region
Autor(es): Luciana Santos Serrão de Castro1; Anderson Nonato do Rosario Marinho2; Elzemar Martins Ribeiro Rodrigues1; Giorgio Christie Tavares Marques3; Tarcísio André Amorim de Carvalho4; Luiz Carlos Santana da Silva5; Sidney Emanuel Batista dos Santos6
DOI: 10.5935/1808-8694.20130016
Palavras-chave: aconselhamento genético; conexinas; surdez.
Keywords: connexins; deafness; genetic counseling.
Resumo:
A deficiência auditiva afeta cerca de 1 em cada 1000 recém-nascidos. Mutações no gene da conexina 26 (GJB2) são as causas mais frequentes de surdez não sindrômica em diferentes populações e é sabido que a mutação delGJB6-D13S1830 em DFNB30 é causadora de surdez neurossensorial. Muitos estudos descrevem o envolvimento de mutações no gene GJB2 com a deficiência auditiva em diferentes populações. Entretanto, existe pouca informação sobre a surdez genética no Brasil, especialmente na região Amazônica.
OBJETIVO: Determinar a prevalência de mutações no gene GJB2 e da mutação delGJB6-D13S1830 em 77 casos esporádicos de surdez não sindrômicas.
MÉTODO: A região codificante do gene GJB2 foi sequenciada e a PCR foi realizada para detectar a mutação delGJB6-D13S1830.
RESULTADOS: O alelo 35delG foi encontrado em 9% dos pacientes (7/77). As mutações M34T e V95M foram detectadas em dois distintos pacientes heterozigotos. A mutação não patogênica V27I foi detectada em 28,6% (22/77). Não foi detectada a mutação delGJB6-D13S1830 em nenhum paciente estudado.
CONCLUSÃO: Alelos mutantes no gene GJB2 foram observados em 40% (31/77) da amostra. Variantes patogênicas foram detectadas em apenas 12% (9/77). Mais estudos são necessários para elucidar causas genéticas de deficiência auditiva em populações miscigenadas.
Abstract:
Hearing impairment affects about 1 in 1000 newborns. Mutations in the connexin 26 (GJB2) gene rank among the most frequent causes of non-syndromic deafness in different populations, while delGJB6-D13S1830 mutation located in the DFNB30 locus is known to cause sensorineural hearing loss. Despite the many studies on the involvement of GJB2 mutations in hearing impairment in different populations, there is little information on genetic deafness in Brazil, especially in the Amazon region.
OBJECTIVE: To determine the prevalence of GJB2 mutations and delGJB6-D13S1830 in 77 sporadic non-syndromic deaf patients.
METHOD: The coding region of the GJB2 gene was sequenced and polymerase chain reaction was performed to detect the delGJB6-D13S1830 mutation.
RESULTS: Mutant allele 35delG was found in 9% of the patients (7/77). Mutations M34T and V95M were detected in two distinct heterozygous patients. Non-pathogenic mutation V27I was detected in 28.6% of the patients (22/77). None of the deaf patients carried the delGJB6-D13S1830 mutation.
CONCLUSION: Mutant alleles on gene GJB2 were observed in 40% (31/77) of the subjects in the sample. Pathogenic variants were detected in only 12% (9/77) of the individuals. More studies are required to elucidate the genetic causes of hearing loss in miscegenated populations.
![]()
INTRODUÇÃO
A surdez afeta aproximadamente um em cada mil nascidos1. A perda auditiva pode estar relacionada a causas genéticas, ambientais ou a uma combinação de ambas. Em países desenvolvidos, cerca de 60% de todos os casos têm origem genética2. Estima-se que 30% dos casos de surdez genética sejam sindrômicos e 70% não sindrômicos3. No Brasil, a frequência de surdez congênita não sindrômica é de cerca de quatro em cada mil nascimentos, 16% dos quais com etiologia genética4. Dentre as manifestações de surdez hereditária não sindrômica, as formas autossômicas recessivas (DFNB) compõem cerca de 75-80% dos casos, as autossômicas dominantes (DFNA) cerca de 20%, as formas ligadas ao X (DFN) de 2-5% e as formas mitocondriais cerca de 1%5. Atualmente, estima-se que mais de 120 diferentes loci estejam envolvidos na surdez e 70 genes já foram identificados e caracterizados6.
O locus GJB2 foi descrito como a principal causa de perda auditiva neurossensorial não sindrômica autossômica recessiva3,7,8. Mutações do gene GJB2 foram associadas a 50% dos casos de perda auditiva não sindrômica autossômica recessiva em muitas populações9,10. Mais de 101 diferentes mutações no gene da conexina 26 foram associadas à deficiência auditiva11. A prevalência de algumas mutações do GJB2 difere consideravelmente entre grupos étnicos. A mutação 35delG é a variante mais comum em populações européias12,13. A frequência do alelo 35delG varia entre 0,97% e 2,24% na região Sudeste do Brasil14,15. No estado de São Paulo, a mutação 35delG foi a mais frequente (12,4%)16; ela foi encontrada em 23% de casos em uma mesma família e 6,2% em casos isolados. A mutação 235delC é geralmente predominante em populações asiáticas17-21, enquanto a mutação 167delT é encontrada com mais frequência em populações de judeus Ashkenazi22-24.
Sabe-se que as mutações do gene GJB6 são uma causa comum de surdez25. A deleção de 243Kb chamada mutação delGJB6-D13S1830 no gene GJB6 é a segunda mais frequente causa genética de surdez pré-lingual não sindrômica na população espanhola26. Ela também já foi descrita em judeus Ashkenazi27 e pacientes franceses com perda auditiva não sindrômica28.
Alguns estudos descreveram a frequência genética das mutações do GJB2 e da mutação delGJB6-D13S1830 associada à surdez não sindrômica em populações brasileiras12,16,29-31. As populações do Norte brasileiro são compostas por uma grande mistura de etnias com a adição de contribuição genética europeia32. Não há dados anteriores disponíveis sobre as variantes alélicas do GJB2 ou a frequência da mutação delGJB6-D13S1830 na região Amazônica. De modo a estabelecer a prevalência das mutações do GJB2 em pacientes com surdez da região, investigamos 77 casos isolados de surdez pré-lingual não sindrômica. Além disso, investigamos a prevalência da deleção delGJB6-D13S1830.
MÉTODO
O presente estudo foi aprovado pelo Comitê de Ética em Pesquisa do Hospital Universitário João de Barros Barreto (protocolo nº 2241/05).
Participantes e avaliação clínica
O estudo foi conduzido com 77 crianças com perda auditiva pré-lingual não sindrômica. Os pacientes incluídos no estudo não tinham qualquer grau de parentesco e representavam casos isolados de surdez. Amostras foram obtidas na Escola para Surdos de Belém, no estado do Pará. Pacientes sindrômicos não foram incluídos no estudo. Todas as 77 crianças tinham deficiência auditiva de grave a profunda. Foi obtido o prontuário médico de cada criança e questionários foram ministrados para garantir que a perda auditiva não tivesse causas ambientais: infecção materno-fetal, complicações pré e perinatais, meningite, caxumba, uso prolongado de antibióticos/medicamentos ototóxicos ou trauma acústico. Todas as crianças foram submetidas à otoscopia, exames audiovestibulares, audiometria e exame geral, incluindo avaliação sistemática de formas sindrômicas de surdez. Os pais de todas as crianças assinaram termo de consentimento informado.
Análise molecular
O DNA foi extraído de sangue completo anticoagulado com EDTA usando o método fenol-clorofórmio-etanol. Para identificar as mutações do GJB2, um fragmento de DNA contendo toda a região codificadora foi amplificado usando o par de primers por Reação em Cadeia da Polimerase (PCR) e dois primers internos foram utilizados no sequenciamento do DNA da Cx26: GJB2-1F (5´-GTGTTGTGTGCATTCGTCTTTTC-3') primer na direção forward para PCR e sequenciamento; GJB2-2R (5´-CCTCATCCCTCTCATGCTGTCTA-3') primer na direção reverse para PCR e sequenciamento; GJB2-4F (5´-GGAAGTTCATCAAGGGGGAGATA-3') primer para sequenciamento interno na direção forward e GJB2-3R (5´-ACCTTCTGGGTTTTGATCTCC TC-3') primer para sequenciamento interno na direção reversa.
As condições do PCR foram 35 ciclos com desnaturação a 94ºC por 1 min, anelamento a 60ºC por 1 min e extensão a 72ºC por 1 min. Em todos os casos da análise do gene GJB2, foi executado sequenciamento bidirecional do DNA e em alguns casos primers internos foram usados para confirmar o resultado. O sequenciamento foi feito com um ABI Prism Big-Dye Terminator Cycle Sequencing KitTM (Applied Biosystems) e a eletroforese com um ABI Prism 377 DNA Sequencer (Applied Biosystems). A amplificação do PCR da mutação delGJB6-D13S1830 foi feita com os primers e segundo as condições anteriormente descritas26.
RESULTADOS
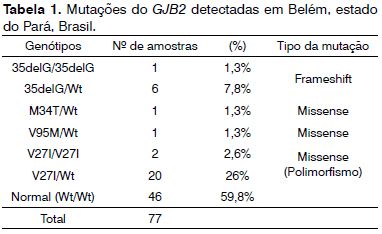
Os resultados encontram-se resumidos na Tabela 1. A análise da região codificadora completa do gene GJB2 das 77 crianças com perda auditiva revelou quatro diferentes mutações já descritas: 35delG, M34T, V95M e V27I. Variantes alélicas no gene GJB2 foram encontradas em 40% dos casos (31 de 77 pacientes). A mutação mais comum foi V27I, detectada em 22 pacientes: vinte indivíduos eram heterozigotos e dois eram homozigotos. Contudo, a mutação V27I já foi descrita como um polimorfismo sem efeitos patológicos não relacionado à perda auditiva17. Mutações patogênicas foram observadas em 12% dos pacientes. A mutação 35delG foi detectada em sete (9%) pacientes: um era homozigoto e seis eram heterozigotos, nos quais a segunda mutação do GJB2 não foi encontrada. A mutação 35delG foi vista em oito dos dez alelos do GJB2 com mutação patogênica. Duas outras mutações missense foram observadas, V95M e M34T, em dois pacientes heterozigotos distintos com o segundo alelo normal. Além disso, a triagem para a mutação del(GJB6-D13S1830) foi negativa para os 77 pacientes.
DISCUSSÃO
O presente estudo é o primeiro a descrever a prevalência das variantes dos genes GJB2 e GJB6 na população amazônica. Vários estudos demonstraram que a mutação 35delG está presente em vários grupos étnicos e geográficos, e que representa até 80% das mutações do gene GJB233. Um estudo anterior descreveu incidência de 2,1% de mutação 35delG na região Norte do Brasil34. Apesar de nosso trabalho ter se baseado num pequeno número de pacientes, seus resultados podem confirmar a importância das mutações do GJB2 na etiologia da perda auditiva em Belém - PA. O presente estudo mostrou que a mutação 35delG é o mais comum alelo patogênico do GJB2. O alelo mutante 35delG foi encontrado em 9% dos pacientes, um achado concordante com estudos feitos anteriormente sobre a população brasileira29,30,35. A relevância patológica da mutação M34T tem sido debatida. Ela foi inicialmente associada à perda auditiva em DFNA38. Entretanto, tal mutação foi descrita por vários autores como sendo polimorfismo, mutação recessiva causal, ou mutação dominante8,36-40. Em um estudo populacional feito no Reino Unido, a M34T se manteve em conformidade com sua classificação como mutação que causa perda auditiva leve/moderada em homozigose ou heterozigose composta. Este achado acrescenta mais evidências de que a M34T é uma variante suave porém funcional com efeito maior no grupo 35delG41. Outro estudo feito na população finlandesa corroborou a hipótese de que a M34T é uma mutação patogênica que exibe um padrão autossômico recessivo de herança associado à perda auditiva neurossensorial não sindrômica suave a moderada em situação de homozigose42.
A mutação M34T foi descrita na população sul-americana43 e brasileira16. A mutação V95M foi observada em apenas um paciente heterozigoto. A mutação missense converte o aminoácido valina em metionina no códon 95. A valina é invariante nessa posição em todos os genes alfa e beta da conexina conhecidos28. A mutação V95M foi descrita como muito mais rara44. Ela já foi relatada em um paciente heterozigoto composto com perda auditiva não sindrômica em uma população do sudeste brasileiro29. De todas as mutações do GJB2 detectadas em nosso estudo, a V27I é a mais frequente. Esta mutação é considerada um polimorfismo e uma alteração não-patológica relacionada à perda auditiva17,22. Ela é raramente observada na população norte-americana22. Contudo, trata-se de uma mutação frequente em populações asiáticas, como a chinesa45; japonesa17; coreana46 e em populações de descendência asiática47. Em um estudo anterior feito com pacientes brasileiros, a mutação V27I foi identificada em dois indivíduos heterozigotos sem parentesco de um grupo de 26 casos esporádicos29.
Dada a elevada prevalência da mutação V27I, consideramos a hipótese da mutação ter resultado da contribuição aumentada dos genes ameríndios das populações do norte brasileiro32. Para abordar esta questão, investigamos a frequência da mutação V27I em uma amostra composta por 400 indivíduos da população de Belém. Os resultados (dados não publicados) demonstraram que a frequência da mutação V27I entre indivíduos com boa audição da população de Belém é alta (12,4%) e semelhante à frequência desta mutação nos pacientes surdos investigados (15,5%). Tais frequências são semelhantes àquelas descritas na China45 e mais elevadas que as da população europeia. Assim, as populações ameríndias provavelmente são a fonte original da mutação V27I na amostra da pesquisa.
A mutação delGJB6-D13S1830 é a segunda mais frequente causa genética de perda auditiva pré-lingual não sindrômica na população espanhola. Foi sugerido um padrão digenético de herança envolvendo mutações do GJB2 e a mutação delGJB6-D13S1830 do gene GJB626. O alelo del(GJB6-D13S1830) também foi identificado em 7,1% dos pacientes com surdez brasileiros26. Em outro estudo, a mutação del(GJB6-D13S1830) foi observada em apenas um paciente heterozigoto composto (35delG/del(GJB6-D13S1830) do Sudeste brasileiro30. No presente trabalho, a triagem dos 77 pacientes não revelou esta mutação e nossos resultados não permitem a atribuição de risco de perda auditiva à mutação del(GJB6-D13S1830) em nossa população.
CONCLUSÃO
Alelos mutantes do GJB2 foram observados em 40% (31/77) dos pacientes surdos amazônicos. Entretanto, variantes patogênicas (35delG, M34T e V95M) -- conhecidas por sua ação recessiva -- foram detectadas em 12% (9/77) da coorte. Dentre os nove pacientes com mutações patogênicas, oito portavam apenas uma mutação na sequência de codificação do GJB2, apesar de toda a região codificadora do gene GJB2 ter sido sequenciada em todas as amostras por sequenciamento bidirecional de DNA, incluindo o heterozigoto 35delG. A segunda mutação putativa presente nesses pacientes poderia estar localizada em áreas não analisadas do gene GJB2, tais como sítios de splice. A hipótese de que a segunda mutação poderia estar localizada em um segundo gene não pode ser descartada.
Os resultados deste estudo piloto deverão contribuir com o desenvolvimento de diagnósticos genéticos para a surdez, desempenhando um importante papel no progresso das questões de saúde pública, no aumento da precisão do aconselhamento genético e no tratamento precoce da região. Entrementes, mais estudos e pesquisas são necessários para descobrir e analisar genes e variantes de modo a elucidar as outras causas genéticas da perda auditiva em populações miscigenadas.
AGRADECIMENTOS
Nossos sinceros agradecimentos aos pacientes por sua participação. O presente trabalho teve o apoio do CNPq.
REFERÊNCIAS
1. Petit C, Levilliers J, Hardelin JP. Molecular genetics of hearing loss. Annu Rev Genet. 2001;35:589-646.
2. Frei K, Szuhai K, Lucas T, Weipoltshammer K, Schöfer C, Ramsebner R, et al. Connexin 26 mutations in cases of sensorineural deafness in eastern Austria. Eur J Hum Genet. 2002;10(7):427-32.
3. Van Camp G, Willems PJ, Smith RJ. Nonsyndromic hearing impairment: unparalleled heterogeneity. Am J Hum Genet. 1997;60(4):758-64.
4. Braga MCC, Otto PA, Spinelli M. Recurrence risks in cases of nonsyndromic deafness. Braz J Dysmorphol Speech Hear Disord. 1999;2:33-40.
5. Smith RJ, Bale JF Jr, White KR. Sensorineural hearing loss in children. Lancet. 2005;5-11;365(9462):879-90.
6. The Hereditary Hearing loss Homepage. [citado 2012 Dec 13]. Disponível em: http://hereditaryhearingloss.org/
7. Gasparini P, Estivill X, Volpini V, Totaro A, Castellvi-Bel S, Govea N, et al. Linkage of DFNB1 to non-syndromic neurosensory autosomal-recessive deafness in Mediterranean families. Eur J Hum Genet. 1997;5(2):83-8.
8. Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387(6628):80-3.
9. Morton CC. Genetics, genomics and gene discovery in the auditory system. Hum Mol Genet. 2002;11(10):1229-40.
10. Rabionet R, Zelante L, López-Bigas N, D'Agruma L, Melchionda S, Restagno G, et al. Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum Genet. 2000;106(1):40-4.
11. The Connexin-deafness Homepage. [citado 2012 Dec 13]. Disponível em: http://davinci.crg.es/deafness/
12. Denoyelle F, Weil D, Maw MA, Wilcox SA, Lench NJ, Allen-Powell DR, et al. Prelingual deafness: high prevalence of a 30delG mutation in the connexin 26 gene. Hum Mol Genet. 1997;6(12):2173-7.
13. Gasparini P, Rabionet R, Barbujani G, Melçhionda S, Petersen M, Brøndum-Nielsen K, et al. High carrier frequency of the 35delG deafness mutation in European populations. Genetic Analysis Consortium of GJB2 35delG. Eur J Hum Genet. 2000;8(1):19-23.
14. Sartorato EL, Gottardi E, de Oliveira CA, Magna LA, Annichino-Bizzacchi JM, Seixas CA, et al. Determination of the frequency of the 35delG allele in Brazilian neonates. Clin Genet. 2000;58(4):339-40.
15. Piatto VB, Oliveira CA, Alexandrino F, Pimpinati CJ, Sartorato EL. Prospects for genetic hearing loss screening: 35delG mutation tracking in a newborn population. J Pediatr (Rio J). 2005;81(2):139-42.
16. Batissoco AC, Abreu-Silva RS, Braga MC, Lezirovitz K, Della-Rosa V, Alfredo T Jr, et al. Prevalence of GJB2 (connexin-26) and GJB6 (connexin-30) mutations in a cohort of 300 Brazilian hearing-impaired individuals: implications for diagnosis and genetic counseling. Ear Hear. 2009;30(1):1-7.
17. Abe S, Usami S, Shinkawa H, Kelley PM, Kimberling WJ. Prevalent connexin 26 gene (GJB2) mutations in Japanese. J Med Genet. 2000;37(1):41-3.
18. Sugata A, Fukushima K, Sugata K, Fukuda S, Kimura N, Gunduz M, et al. High-throughput screening for GJB2 mutations--its clinical application to genetic testing in prelingual deafness screening for GJB2 mutations. Auris Nasus Larynx. 2002;29(3):231-9.
19. Wang YC, Kung CY, Su MC, Su CC, Hsu HM, Tsai CC, et al. Mutations of Cx26 gene (GJB2) for prelingual deafness in Taiwan. Eur J Hum Genet. 2002;10(8):495-8.
20. Liu XZ, Xia XJ, Ke XM, Ouyang XM, Du LL, Liu YH, et al. The prevalence of connexin 26 (GJB2) mutations in the Chinese population. Hum Genet. 2002;111(4-5):394-7.
21. Ohtsuka A, Yuge I, Kimura S, Namba A, Abe S, Van Laer L, et al. GJB2 deafness gene shows a specific spectrum of mutations in Japan, including a frequent founder mutation. Hum Genet. 2003;112(4):329-33.
22. Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, et al. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am J Hum Genet. 1998;62(4):792-9.
23. Morell RJ, Kim HJ, Hood LJ, Goforth L, Friderici K, Fisher R, et al. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med. 1998;339(21):1500-5.
24. Sobe T, Erlich P, Berry A, Korostichevsky M, Vreugde S, Avraham KB, et al. High frequency of the deafness-associated 167delT mutation in the connexin 26 (GJB2) gene in Israeli Ashkenazim. Am J Med Genet. 1999;86(5):499-500.
25. Grifa A, Wagner CA, D'Ambrosio L, Melchionda S, Bernardi F, Lopez-Bigas N, et al. Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat Genet. 1999;23(1):16-8.
26. del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Tellería D, et al. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med. 2002;346(4):243-9.
27. Lerer I, Sagi M, Ben-Neriah Z, Wang T, Levi H, Abeliovich D. A deletion mutation in GJB6 cooperating with a GJB2 mutation in trans in non-syndromic deafness: A novel founder mutation in Ashkenazi Jews. Hum Mutat. 2001;18(5):460.
28. Pallares-Ruiz N, Blanchet P, Mondain M, Claustres M, Roux AF. A large deletion including most of GJB6 in recessive non syndromic deafness: a digenic effect? Eur J Hum Genet. 2002;10(1):72-6.
29. Oliveira CA, Maciel-Guerra AT, Sartorato EL. Deafness resulting from mutations in the GJB2 (connexin 26) gene in Brazilian patients. Clin Genet. 2002;61(5):354-8.
30. Belintani Piatto V, Maria Goloni Bertollo E, Lúcia Sartorato E, Victor Maniglia J. Prevalence of the GJB2 mutations and the del (GJB6-D13S1830) mutation in Brazilian patients with deafness. Hear Res. 2004;196(1-2):87-93.
31. Cordeiro-Silva Mde F, Barbosa A, Santiago M, Provetti M, Rabbi-Bortolini E. Prevalence of 35delG/GJB2 and del (GJB6-D13S1830) mutations in patients with non-syndromic deafness from a population of Espírito Santo-Brazil. Braz J Otorhinolaryngol. 2010;76(4):428-32.
32. Santos SEB, Guerreiro JF. The indigenous contribution to the formation of the population of the Brazilian Amazon region. Rev Bras Genet. 1995;18:311-5.
33. Denoyelle F, Marlin S, Weil D, Moatti L, Chauvin P, Garabédian EN, et al. Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin-26 gene defect: implications for genetic counselling. Lancet. 1999;353(9161):1298-303.
34. Oliveira CA, Pimpinati CJ, Alexandrino F, Magna LA, Maciel-Guerra AT, Sartorato EL. Allelic frequencies of the 35delG mutation of the GJB2 gene in different Brazilian regions. Genet Test. 2007;11(1):1-3.
35. da Motta LH, Félix TM, de Souza LT, Lavinsky-Wolff M, Costa-Motta FM, de Faria MR, et al. Prevalence of the 35delG mutation in deaf South Brazilian infants submitted to cochlear implantation. Int J Pediatr Otorhinolaryngol. 2012;76(2):287-90.
36. Scott DA, Kraft ML, Stone EM, Sheffield VC, Smith RJ. Connexin mutations and hearing loss. Nature. 1998;391(6662):32.
37. Wilcox SA, Saunders K, Osborn AH, Arnold A, Wunderlich J, Kelly T, et al. High frequency hearing loss correlated with mutations in the GJB2 gene. Hum Genet. 2000;106(4):399-405.
38. Griffith AJ, Chowdhry AA, Kurima K, Hood LJ, Keats B, Berlin CI, et al. Autosomal recessive nonsyndromic neurosensory deafness at DFNB1 not associated with the compound-heterozygous GJB2 (connexin 26) genotype M34T/167delT. Am J Hum Genet. 2000;67(3):745-9.
39. Zoll B, Petersen L, Lange K, Gabriel P, Kiese-Himmel C, Rausch P, et al. Evaluation of Cx26/GJB2 in German hearing impaired persons: mutation spectrum and detection of disequilibrium between M34T (c.101T>C) and -493del10. Hum Mutat. 2003;21(1):98.
40. Feldmann D, Denoyelle F, Loundon N, Weil D, Garabedian EN, Couderc R, et al. Clinical evidence of the nonpathogenic nature of the M34T variant in the connexin 26 gene. Eur J Hum Genet. 2004;12(4):279-84.
41. Hall A, Pembrey M, Lutman M, Steer C, Bitner-Glindzicz M. Prevalence and audiological features in carriers of GJB2 mutations, c.35delG and c.101T>C (p.M34T), in a UK population study. BMJ Open. 2012;2(4). pii: e001238.
42. Löppönen T, Dietz A, Väisänen ML, Valtonen H, Kosunen A, Hyvärinen A, et al. Homozygous M34T mutation of the GJB2 gene associates with an autosomal recessive nonsyndromic sensorineural hearing impairment in Finnish families. Acta Otolaryngol. 2012;132(8):862-73.
43. Dalamón V, Béhèran A, Diamante F, Pallares N, Diamante V, Elgoyhen AB. Prevalence of GJB2 mutations and the del(GJB6-D13S1830) in Argentinean non-syndromic deaf patients. Hear Res. 2005;207(1-2):43-9.
44. Cucci RA, Prasad S, Kelley PM, Green GE, Storm K, Willocx S, et al. The M34T allele variant of connexin 26. Genet Test. 2000;4(4):335-44.
45. Shi GZ, Gong LX, Xu XH, Nie WY, Lin Q, Qi YS. GJB2 gene mutations in newborns with non-syndromic hearing impairment in Northern China. Hear Res. 2004;197(1-2):19-23.
46. Park HJ, Hahn SH, Chun YM, Park K, Kim HN. Connexin26 mutations associated with nonsyndromic hearing loss. Laryngoscope. 2000;110(9):1535-8.
47. Posukh O, Pallares-Ruiz N, Tadinova V, Osipova L, Claustres M, Roux AF. First molecular screening of deafness in the Altai Republic population. BMC Med Genet. 2005;6:12.
1. Doutorado em Genética e Biologia Molecular (Pesquisadora).
2. Doutorado em Genética e Biologia Molecular (Pesquisador).
3. Graduação em Biomedicina.
4. Mestrado em Genética e Biologia Molecular (Pesquisador).
5. Doutorado (Professor Associado I - Instituto Ciencias Biológicas - Universidade Federal do Pará).
6. Doutorado (Professor Associado III - Instituto Ciencias Biológicas - Universidade Federal do Pará).
Instituto de Ciências Biológicas - Universidade Federal do Pará.
Endereço para correspondência:
Luciana Santos Serrão de Castro
Tv. 14 de Abril, nº 1235, apto 601
Belém - PA. Brasil. CEP: 66060-460.
CNPq-Brazilian National Research Council.
Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) do BJORL em 16 de outubro de 2012. cod. 10524.
Artigo aceito em 2 de novembro de 2012.
Imprimir: ![]()