

Ano: 2011 Vol. 77 Ed. 6 - Novembro - Dezembro - (16º)
Seção: Artigo Original
Páginas: 784 a 791
 PDF PT
PDF PT Estudo audiológico e genético de lactentes de alto risco
Audiological and genetics studies in high-risk infants
Autor(es): Maria Francisca Colella-Santos1; Maria de Fátima de Campos Françozo2; Christiane Marques do Couto3; Maria Cecilia Marconi Pinheiro Lima4; Tatiana Guilhermino Tazinazzio5; Arthur Menino Castilho6; Edi Lucia Sartorato7
Palavras-chave: audição, lactente perda auditiva, testes auditivos.
Keywords: child, hearing, hearing loss, hearing tests.
Resumo:
A audição é um dos principais meios de contato do indivíduo com o mundo externo, desempenhando papel fundamental na integração com a sociedade. Objetivo: Analisar os resultados obtidos na avaliação audiológica, otorrinolaringológica e genética de lactentes de alto risco que falharam na triagem auditiva neonatal. Material e Método: Estudo clínico e experimental.Foram avaliados 38 lactentes, entre 1 e 6 meses de idade cronológica. Os procedimentos utilizados foram: anamnese, imitanciometria, Potencial Evocado Auditivo de Tronco Encefálico, Emissões Otoacústicas por Transiente e avaliação otorrinolaringológica. O estudo genético foi realizado a partir da extração de DNA da mucosa bucal utilizando o método de protocolo adaptado no Laboratório de Genética Humana do CBMEG/UNICAMP. Resultado: Não houve diferença estatisticamente significante entre neonatos com audição normal e perda auditiva e as variáveis gênero e número de indicadores de risco. Quanto à idade gestacional, neonatos a termo foram mais afetados. A perda auditiva esteve presente em 58% da amostra, sendo do tipo condutiva em 31,5%, e neurossensorial em 28,9% dos casos. Não foram encontradas as mutações genéticas mais comumente observadas em casos com etiologia genética. Conclusão: A perda auditiva foi diagnosticada na maioria dos lactentes de risco com provável etiologia ambiental.
Abstract:
Hearing is one of the main ways with which one person can contact the external world; it plays a key role in their integration with society. Aim: The objective of this study was to analyze the results of the hearing, medical and genetic evaluation of high-risk infants who failed the newborn hearing screening. Materials and Methods: Clinical and experimental study. We assessed thirty-eight neonates, with ages between one and six months. The infants underwent the following procedures: medical interview; immittance testing; Brainstem Auditory Evoked Potential; Transient Evoked Otoacoustic Emission and otorhinolaryngological evaluation. DNA extraction from the oral mucosa was performed for genetic studies using the protocol method adapted from the Human Genetics Lab of the CBMEG/UNICAMP. Results: Regarding gender and presence of risk factors, significant statistically differences were not found in normal hearing infants and in those with hearing loss. Concerning gestational age, term infants were more affected by hearing loss. Hearing loss was identified in 58% of the sample, conduction hearing loss represented 31.5% (12/38) and neurossensory 28.9% of cases.There were none of the genetic mutations most commonly seen in cases with a genetic etiology. Conclusion: Hearing loss was identified in the majority of High-risk infants.
![]()
INTRODUÇÃO
A audição é um dos principais meios de contato do indivíduo com o mundo externo, desempenhando papel fundamental na sua integração com a sociedade.
Sabe-se que a incidência de perda auditiva bilateral e significante é estimada entre um a três a cada mil recém-nascidos (RNs) de baixo risco para perda auditiva1,2, aumentando para cerca de dois a cinco em cada 100 RNs provenientes de Unidades de Terapia Intensiva (UTI)1,3,4, ou 10,2%, como encontrado por Lima et al.5,6. De acordo com o Instituto Brasileiro de Geografia e Estatística, no Censo realizado em 2000, a incidência de perda auditiva na população brasileira foi de 16,7%, sendo de 16,4% no Estado de São Paulo7. A perda auditiva é a alteração congênita mais frequente, com maior prevalência do que outras doenças rotineiramente triadas, sendo 100 vezes mais prevalente do que a fenilcetonúria e 10 vezes mais prevalente que o hipotireoidismo8. A surdez infantil é considerada um verdadeiro problema de saúde pública, devido à sua elevada prevalência e às múltiplas consequências que acarreta, afetando não apenas o desenvolvimento da linguagem, mas também o desenvolvimento cognitivo, intelectual, cultural e social da criança9,10.
A triagem auditiva neonatal universal tem sido recomendada como principal estratégia para diminuir a idade em que o diagnóstico da perda auditiva é realizado11,12.É a primeira etapa de um programa de saúde auditiva neonatal, devendo ser seguida pelo atendimento multidisciplinar para diagnóstico, envolvendo principalmente o fonoaudiólogo, o médico otorrinolaringologista e, em alguns serviços, o geneticista. Após o diagnóstico, devem ser iniciados os processos de intervenção, com uso de amplificação sonora e reabilitação. O diagnóstico precoce, seguido por medidas de intervenção médica e fonoaudiológica, permitirá o contato da criança com o mundo sonoro ainda no período de grande plasticidade do sistema nervoso central, o primeiro ano de vida, permitindo o aumento das conexões nervosas e possibilitando melhores resultados na reabilitação auditiva e desenvolvimento geral da criança acometida pela perda auditiva13,14.
Para o diagnóstico da perda auditiva, é recomendado que se realize uma bateria de testes, utilizando avaliações comportamentais e eletrofisiológicas, tais como: observação do comportamento auditivo a sons calibrados e não calibrados, medidas de imitância acústica, emissão otoacústica, Potencial Evocado Auditivo de Tronco Encefálico-PEATE. Esta bateria de testes é sugerida para determinar o tipo, grau e configuração da perda auditiva, a fim de realizar o diagnóstico diferencial, a intervenção e a indicação de próteses auditivas em alterações neurossensoriais, mistas ou condutivas15. A pesquisa etiológica da surdez é de extrema importância para que o prognóstico seja estabelecido, assim como as condutas apropriadas.
Várias são as causas da perda auditiva congênita, ou seja, quando adquirida no período pré-natal ou nos primeiros dias após o nascimento. Podemos classificar as causas da perda auditiva em genéticas e ambientais.A etiologia de origem genética pode ser sindrômica, ou seja, apresentar-se associada a malformações craniofaciais ou cervicais, displasias esqueléticas, anomalias cutâneas ou oculares, doenças neurológicas e disfunções renais ou metabólicas, entre outras. Estima-se que 30% dos casos de surdez pré-lingual sejam sindrômicos e 70% não-sindrômicos16. A surdez não-sindrômica pode se apresentar em vários padrões de herança: ligadas ao cromossomo X (DFN), em 1%-3% dos casos; em formas autossômicas dominantes (DFNA), em 15%; e em formas autossômicas recessivas (DFNB) em 80%. Além disso, existem os casos de herança materna, devido a mutações em genes mitocondriais16.Nos países desenvolvidos, em torno de 60% dos casos de perda auditiva são de origem genética17.
Os fatores ambientais incluem, entre outros, infecções congênitas, fatores perinatais e pós-natais.
Assim, o objetivo deste estudo foi analisar os resultados obtidos na avaliação audiológica, otorrinolaringológica e genética de lactentes de alto risco que falharam na triagem auditiva neonatal, considerando as variáveis sexo masculino e feminino, número de indicadores de risco e idade gestacional. Além disso, classificar a perda auditiva quanto ao tipo, grau, lado afetado e possível etiologia.
MATERIAL E MÉTODOS
Estudo de coorte contemporânea transversal, aprovado pelo Comitê de Ética e Pesquisa da Faculdade de Ciências Médicas-FCM/UNICAMP, sob protocolo no 028/2008.
Foram incluídos no estudo todos os lactentes nascidos no Hospital da Mulher Prof. Dr. José Aristodemo Pinotti - CAISM/UNICAMP, que permaneceram na UTI neonatal e que falharam na triagem auditiva, no período de fevereiro de 2009 a março de 2010. Foram excluídas as crianças nascidas em outro serviço de saúde da região ou que não concluíram todas as avaliações no período do estudo. Foram encaminhados para avaliação diagnóstica 52 lactentes, número este próximo ao cálculo amostral baseado na média mensal de partos que o hospital realiza, 250, além do número de recém-nascidos que ficam internados em UTI, 40 mensais. Assim sendo, houve 500 recém-nascidos em 12 meses, sendo que, destes, em média 10% falham na triagem auditiva6, ou seja, 50 lactentes. O teste de triagem auditiva utilizado foi Potencial Evocado Auditivo de Tronco Encefálico Automático-PEATE-A. O equipamento utilizado foi Algo 2e color- NATUS. Consideramos que o lactente PASSOU na triagem auditiva quando apresentou resposta para 35 dB bilateralmente no PEATE-A.
A avaliação audiológica foi realizada por fonoaudióloga da equipe no Laboratório de Diagnóstico Audiológico do CEPRE/FCM/unicamp, entre 1 e 6 meses de idade cronológica da criança, em ambiente acusticamente tratado. Foi constituída pelos procedimentos: anamnese, avaliação das condições da orelha média, aplicação do PEATE (pesquisa do limiar eletrofisiológico e da integridade da via auditiva) e das EOAT. Durante a aplicação destes procedimentos, a criança estava em sono natural.
A anamnese foi realizada com os familiares, registrando-se dados de identificação, dados obtidos no relatório de alta do lactente, além de informações sobre o desenvolvimento motor, auditivo e de linguagem. Foram considerados indicadores de risco para perda auditiva os recomendados pelo JCIH18, que também foram coletados a partir do relatório de alta elaborado pela equipe de neonatologistas.
O limiar eletrofisiológico, assim como a integridade da via auditiva, foram avaliados com o uso do PEATE captado pelo equipamento Eclipse EP 25 - Interacoustics, com fones de inserção. A pesquisa da integridade da via auditiva foi feita utilizando-se estímulo do tipo clique a 80 dB, com velocidade de apresentação do estímulo de 19 estímulos/segundo, não variável, que permitiu a avaliação da via auditiva até o tronco encefálico e a identificação de possíveis alterações neste trajeto. O limiar eletrofisiológico foi obtido com estímulos descendentes, até chegarmos à menor intensidade do estímulo que desencadeou o aparecimento da onda V. A estimulação foi repetida duas vezes, para verificar a reprodutibilidade do traçado e garantir a presença de resposta. Para captação das respostas, foram fixados eletrodos de superfície com pasta eletrolítica nas mastoides direita e esquerda do lactente e na posição frontoparietal, após limpeza do local com pasta abrasiva.Foram analisados os seguintes parâmetros: presença das ondas I, III e V; latência absoluta de ondas I, III e V; latências interpicos I-V, I-III, III-V; amplitude da onda V em relação à amplitude da onda I; diferença interaural da latência interpico I-V ou da latência da onda V. As EOAT foram captadas por meio do equipamento ILO 292 USBII.
As condições de orelha média foram avaliadas por meio da curva timpanométrica, com tom de sonda de 1000 Hz e pesquisa do reflexo acústico ipsilateral nas frequências de 500 a 4000 Hz. O equipamento utilizado foi o 235 H, Interacoustics.
O registro das respostas obtidas em cada teste foi realizado em folhas de respostas. Consideramos audição normal, limiar eletrofisiológico para cliques menor que 30 dB e latências absolutas e interpicos dentro dos valores esperados para a idade gestacional19, além de presença de emissões otoacústicas por transiente20, curva timpanométrica do tipo A e presença de reflexo acústico ipsilateral21,22.
Os lactentes que apresentaram resultados alterados em pelo menos um teste auditivo foram encaminhados para avaliação otorrinolaringológica, sob supervisão do profissional da equipe, que foi constituída pelo exame otoscópico para verificar as condições do meato acústico externo e da membrana timpânica, além de exames de imagens solicitados quando necessário.
A partir da análise conjunta entre avaliação audiológica e otorrinolaringológica, classificamos os resultados em audição normal ou perda auditiva. A perda auditiva foi classificada considerando-se o tipo23 e o grau segundo Silman & Silverman24 e em unilateral ou bilateral. O rastreamento genético foi realizado por meio da extração de DNA da mucosa bucal, colhido pela examinadora após realização dos testes auditivos, em todos que falharam na triagem auditiva. A partir do DNA obtido de células da mucosa bucal, a mutação 35delG foi analisada pela técnica de AS-PCR padronizada pelo laboratório de Genética Molecular Humana do CBMEG. (patente no P10005340-6; Método de teste para surdez de origem genética). Por meio da técnica de PCR, foram analisadas as deleções D(GJB6- D13S1830) e D(GJB6-D13S1854) utilizando primers previamente descritos25. Foi realizado um único teste diagnóstico envolvendo as duas deleções em um mesmo PCR. Para análise das mutações mitocondriais, foram amplificados fragmentos de DNAmt do gene MTRNR1, para detectar a mutação A1555G, utilizando pares de primers previamente descritos. Os produtos de amplificação foram submetidos à análise de restrição para detecção das mutações.
A análise estatística foi realizada por meio do software SAS versão 9.1.3. O nível de significância assumido foi de 5% e foi assinalado com asterisco (*).
RESULTADOS
Foram encaminhados para avaliação diagnóstica 52 lactentes que falharam na triagem auditiva neonatal. Destes neonatos, participaram do estudo 38 (73%) que concluíram todas as etapas do processo diagnóstico. As demais crianças apresentaram faltas nos dias agendados, não dormiram para a realização do PEATE, dentre outros fatores que dificultaram a conclusão do processo diagnóstico no período deste estudo.
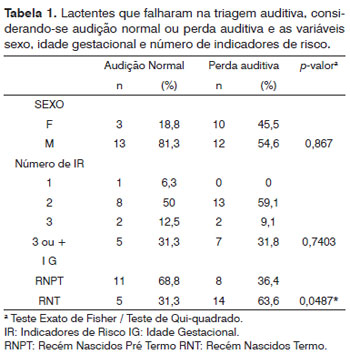
Apresentam-se na Tabela 1 os resultados obtidos para lactentes com audição normal ou perda auditiva, considerando-se as variáveis sexo masculino e feminino, número de indicadores presentes na história clínica e idade gestacional.
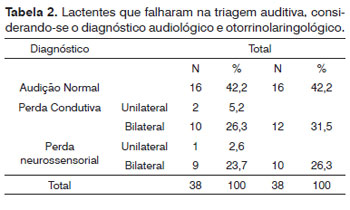
Na Tabela 2, pode-se observar a distribuição dos lactentes avaliados, considerando-se o diagnóstico audiológico e otorrinolaringológico.
No estudo genético, não foi encontrada a mutação 35delG no gene da conexina 26 (GJB2), assim como as deleções D(GJB6-D13S1830) e Δ(GJB6-D13S1854) no gene GJB6 e a mutação A1555G presentes no gene mitocondrial MTRNR.
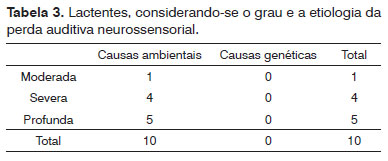
Na Tabela 3, mostram-se os lactentes com perda auditiva neurossensorial, considerando-se o grau e possível etiologia.
DISCUSSÃO
A triagem auditiva neonatal é o principal meio de detectar precocemente perdas auditivas. O procedimento deve ser rápido, simples e selecionar aqueles com maior probabilidade de uma alteração na função testada26. Os recém-nascidos (RNs) internados em UTI Neonatal do Caism/UNICAMP foram submetidos à triagem auditiva, preferencialmente antes da alta hospitalar, por meio de procedimentos automáticos. O PEATE - A tem sido recomendado principalmente para os RNs que permaneceram em unidades de terapia intensiva, devido à maior ocorrência nesta população de indicadores de risco para alterações retrococleares, que envolvem as células ciliadas internas, vias auditivas e/ou tronco encefálico18,27.
Falharam na triagem auditiva 38 lactentes, sendo que 25 (66%) dos neonatos eram do sexo masculino e 50% eram prematuros. Verificamos que a maioria das crianças estudadas eram do sexo masculino, o que reflete a distribuição demográfica do gênero próprio da maioria das UTIs neonatais28.
Com relação aos indicadores de risco presentes na história clínica, todos apresentaram pelo menos um indicador de risco (Tabela 1).
Verificamos que não houve diferença estatisticamente significativa entre neonatos com audição normal e perda auditiva e as variáveis gênero e número de indicadores de risco. No entanto, ao considerarmos a idade gestacional, neonatos a termo foram mais afetados pela perda auditiva do que os prematuros, sendo a diferença estatisticamente significativa (Tabela 1). Os neonatos nascidos a termo deste estudo necessitaram de cuidados intensivos por apresentarem intercorrências graves ao nascimento, como anoxia, malformações congênitas, síndromes, infecção congênita, todas indicadores de risco para perda auditiva. A literatura apresenta características neonatais para o grupo de crianças com perda auditiva diagnosticada a partir de um programa de triagem auditiva neonatal, que incluem idade gestacional maior que 37 semanas e peso ao nascimento maior que 2500 gramas28.
A partir da análise conjunta dos resultados dos testes auditivos aplicados, constatamos que apresentaram resultados normais, em todos os testes aplicados, 42% (16/38) avaliadas (Tabela 2). O fato de terem falhado na triagem auditiva e apresentarem resultados normais nos testes auditivos aplicados pode sugerir uma alteração temporária no sistema auditivo do tipo condutiva, que foi recuperada ou então atraso na mielinização das vias auditivas até o tronco encefálico. As crianças que apresentaram indicadores de risco em suas histórias clínicas, que podem estar relacionados à perda auditiva progressiva e/ou de aparecimento tardio, foram encaminhadas para monitoramento do desenvolvimento auditivo e de linguagem.
A perda auditiva condutiva esteve presente em 31,5% (12/38) da amostra, sendo bilateral na maioria dos casos (Tabela 2). Com relação às crianças com perda auditiva, o tipo condutivo ocorreu em 55% (12/22) das crianças.Nos testes audiológicos, evidenciamos ausência de emissões otoacústicas por transiente acompanhada pela curva timpanométrica do tipo B e ausência de reflexos acústicos ipsilaterais, além da integridade de via auditiva para 80 dB no PEATE. Verificamos que em 4/12 (33,33%) dos casos condutivos, a Síndrome de Down estava presente e em um dos casos condutivos unilaterais havia má-formação da orelha externa e do meato acústico externo.
Resultados semelhantes foram obtidos por Bone et al.29, que encontraram alterações condutivas em crianças que falharam na triagem auditiva, sendo a otite média a etiologia mais comum nos casos estudados. Caracterizase por ser variável, episódica, podendo variar de grau leve a moderado, não ultrapassando a 50 dB30. É uma doença altamente prevalente na infância, especialmente em crianças de alto risco, sendo que o 4º e o 12º meses são os de maior ocorrência de secreção na orelha média e os de menor ocorrência são os três primeiros meses de vida30-33. É multifatorial e em lactentes podemos destacar o aleitamento materno ausente ou desmame precoce, posição deitada ao mamar, primeiro episódio de otite média aguda antes dos seis meses de idade, imaturidade ou deficiência imunológica, permanência em creches ou berçários com aumento do número de gripes e resfriados, tabagismo passivo34. Crianças que apresentam otites secretoras no período neonatal são de maior risco para otite média crônica durante o primeiro ano de vida35. Estudando a ocorrência e recorrência de secreção de orelha média, verificaram que houve maior recorrência de quatro ou mais episódios para o sexo masculino e o período de maior incidência de secreção na orelha média foi o primeiro ano de vida. Encontraram também que crianças que foram amamentadas até o 6º mês de vida apresentaram maior recorrência de quatro ou mais episódios. O inverso foi encontrado para os lactentes amamentados acima de 10 meses. Os autores sugerem a implantação de programas que objetivem prevenção, diagnóstico e tratamento das otites, principalmente pelo fato dos primeiros anos de vida serem fundamentais para o desenvolvimento, além do incentivo pelos profissionais da saúde para o aleitamento materno30.
Medidas preventivas, como incentivo à amamentação natural e o posicionamento adequado durante as mamadas, assim como tabagismo passivo diminuído, dentre outras, podem minimizar as alterações auditivas condutivas, que podem levar a um prejuízo considerável no desenvolvimento das habilidades auditivas e de linguagem decorrente da privação sensorial e a flutuação da audição própria das otites médias.
Nas crianças com Síndrome de Down (SD), a incidência da perda auditiva varia de 2,6% a 67,5%, sendo atribuída principalmente à alta incidência de orelha média secretora36,37. Strome et al.37 encontraram 70% de efusão de orelha média de 107 pacientes com SD e idade inferior a 1 ano. Diversos fatores parecem predispor à ocorrência de orelha média secretora: anormalidade anatômica da tuba auditiva e da cadeia ossicular, disfunção dos músculos responsáveis pela abertura da tuba auditiva e estenose do meato acústico externo37.
Estas crianças encontravam-se em tratamento otorrinolaringológico e reavaliações audiológicas periódicas, assim como acompanhamento do desenvolvimento auditivo e de linguagem. Há relatos da literatura referindo que em alguns casos de orelha média há melhora espontânea sem ocorrer danos ao desenvolvimento. Em outros, há risco de a doença se tornar crônica, podendo levar a distúrbios do desenvolvimento da linguagem e educacional, além do que se não tratada, pode evoluir, agravar-se e comprometer as células mastoideas, ou mesmo a cavidade craniana, causando graves complicações e até afetar a orelha interna, provocando perda de audição neurossensorial29,33.
Verificamos perda auditiva neurossensorial em 28,9% (10/38) da amostra, sendo cinco casos de grau variando de moderado a grave e cinco casos de grau profundo (Tabela 3). Estas crianças foram encaminhadas para indicação e adaptação de prótese auditiva, orientação aos pais e reabilitação fonoaudiológica. Os indicadores de risco mais frequentes presentes nas histórias clínicas foram hiperbilirrubinemia, anoxia perinatal e ventilação mecânica, além da permanência em UTI por mais que 5 dias.
Dentre os casos de perda auditiva neurossensorial profunda bilateral, três crianças apresentaram resultados compatíveis com espectro da neuropatia auditiva (ENA), ou seja, presença de microfonismo coclear e ausência de ondas I, III e V a 100 dB no PEATE e EOAT presentes, além de curva timpanométrica do tipo A e ausência de reflexos acústicos. Nos três casos, constatamos a presença de hiperbilirrubinemia como indicador de risco, com Bilirrubina Total maior que 28 mg/dl.
Características do espectro da neuropatia auditiva (ENA) incluem a dessincronia do VIII par craniano e/ ou tronco encefálico, um distúrbio das células ciliadas internas, uma disfunção das fibras do gânglio espiral38 ou uma alteração na transmissão aferente na sinapse entre as células ciliadas internas e o VIII par craniano39 e função normal das células ciliadas externas. A maioria dos casos tem alteração bilateral, variando de grau grave a profundo. No entanto, casos unilaterais e perdas moderadas foram descritas, o que revela uma entidade não homogênea40. A ENA pode ocorrer na ausência de qualquer outra condição médica aparente. No entanto, uma história perinatal de hiperbilirrubinemia é frequente, além da asfixia ou anoxia40. O ENA é muito mais comum em crianças que permaneceram em UTI neonatal40. A literatura aponta uma prevalência, que varia entre 0,2% até 4%, no grupo de crianças de risco para perda auditiva e de 0,5%-15% entre crianças com perda auditiva conhecida40,41. Estudos que incluem crianças maiores ou que frequentam escolas de surdos apresentam prevalência muito menor, possivelmente pelo fato de que a ENA só é diagnosticada de forma objetiva nos meses iniciais de vida em que as emissões otoacústicas estão presentes pela função normal das células ciliadas externas. Com a evolução da doença, estas células não apresentam mais atividade ou podem ter sido lesadas por amplificação sonora das próteses auditivas em crianças não diagnosticadas. Declau et al.17 estabeleceram como diagnóstico do ENA para dois casos (4,2%), sendo um causado pela hiperbilirrubinemia no período perinatal e o outro sem indicadores de risco presentes. O diagnóstico diferencial entre as perdas neurossensoriais é importante, já que os procedimentos terapêuticos diferem das outras formas de perda auditiva permanente.
No estudo genético, todos os indivíduos avaliados foram normais para a mutação 35delG do gene da conexina 26 (GJB2). No gene GJB2 foi encontrado o polimorfismo V27I no individuo 1 em heterozigose, assim como as mutações silenciosas, que apresentam pouca relevância para a perda auditiva. Apesar de não ter sido encontrada em nenhum dos indivíduos, o rastreamento da mutação 35delG é de extrema importância, visto que está presente em 70% dos casos de surdez quando há o envolvimento do gene GJB2. No Brasil, foi determinada a prevalência de 0,97% de portadores da mutação 35delG, aproximadamente 1:103 heterozigotos, em um rastreamento realizado em 620 neonatos, na região de Campinas, SP42. O resultado negativo de mutações no gene GJB2 diminui o risco empírico relacionado à etiologia genética da surdez. Além do gene GJB2, também foram analisadas as deleções Δ(GJB6-D13S1830) e Δ(GJB6-D13S1854) encontradas no gene GJB6. No presente trabalho, não foi encontrado nenhum indivíduo com estas deleções. Também foi estudada a mutação mitocondrial A1555G associada à perda de audição e uso de antibióticos aminoglicosídeos. Essa alteração não foi encontrada em nenhum dos indivíduos estudados.
Declau et al.17 realizaram um estudo prospectivo com o objetivo de analisar os achados audiológicos e causas da perda auditiva de 170 crianças que falharam na triagem auditiva neonatal, sendo que 13 permaneceram em UTI neonatal. As crianças que falharam foram encaminhadas para avaliação por meio de testes eletrofisiológicos, como PEATE, resposta de estado estável e/ou testes comportamentais.As crianças provenientes de UTI neonatal apresentaram perda auditiva permanente em 61,5% dos casos. A razão de perda auditiva considerando-se o sexo masculino/feminino foi de 3/0. A média da perda auditiva foi de 60 dBNA. Os fatores de risco mais prevalentes foram ventilação mecânica, baixo peso e hiperbilirrubinemia. Quanto à etiologia, 39,6% dos casos de perda auditiva foram atribuídos a causas ambientais, sendo a mais frequente a infecção congênita por Citomegalovirus (18,8% dos casos).
Quanto à etiologia da perda auditiva, a literatura aponta que, em países desenvolvidos, em 50% dos casos a origem da perda auditiva é genética e em 50% a causa é ambiental42,43. De acordo com a literatura, os mais importantes fatores ambientais relacionados à perda auditiva neurossensorial são infecção congênita, ototoxicidade, prematuridade e anoxia neonatal43.
No nosso estudo, o mais provável é que os fatores ambientais presentes na história clínica das crianças tenham sido a causa da perda auditiva, uma vez que não foram encontradas as mutações genéticas mais comumente observadas em casos com etiologia genética (Tabela 3). A identificação da etiologia da perda auditiva é um aspecto relevante que fornece novas informações para a reabilitação auditiva, o prognóstico para a criança e a família. Além disso, estes estudos podem colaborar para esclarecer fatores epidemiológicos da perda auditiva, o que contribuirá para o planejamento de ações preventivas e programas de vigilância
CONCLUSÃO
A partir da análise dos resultados obtidos nos achados audiológicos, otorrinolaringológicos e genéticos de lactentes de alto risco que falharam na triagem auditiva, conclui-se que a perda auditiva foi mais frequente em neonatos a termo do que em prematuros, houve uma distribuição semelhante de crianças com perda auditiva do tipo condutiva e neurossensorial predominantemente bilateral. A provável etiologia das perdas neurossensoriais deveu-se a fatores ambientais. O diagnóstico e a pesquisa etiológica da surdez são de extrema importância para que o prognóstico seja estabelecido, assim como as condutas apropriadas.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Knott C. Universal newborn hearing screening coming soon: "hear's" why. Neonatal Netw. 2001;20(8):25-33.
2. Kennedy C, McCann D. Universal neonatal hearing screening moving from evidence to practice. Arch Dis Child Fetal Neonatal Ed. 2004;89(5):F378-83.
3. Northern JL, Hayes D. Universal screening for infant hearing impairment: necessary, beneficial, and justifiable. Audiol Today. 1994;6(2):10-3.
4. Durante AS, Carvalho RMM, Costa MTZ, Cianciarullo MA, Voegels RL, Takahashi GM, et al. Programa de Triagem Auditiva Neonatal - Modelo de Implementação. Arq Int Otorrinolaringol. 2004;8(1):40-6.
5. Lima GML, Marba ST, Santos MFC. Avaliação auditiva em recémnascidos internados em Unidade de Terapia Intensiva e de Cuidados Intermediários: triagem e acompanhamento ambulatorial. Rev Cienc Med. 2005;14(2):147-56.
6. Lima GML, Marba ST, Santos MFC. Avaliação auditiva em recém-nascidos internados em UTI Neonatal. J Pediatr (Rio J). 2006;82(2):110-4.
7. Instituto Brasileiro de Geografia e Estatística. Censo de 2000. Disponível em http://www.ibge.gov.br/censo/default.php 17 março 2010 8. Baroch KA. Universal newborn hearing screening: finetuning process. Curr Opin Otolaryngol Head Neck Surg. 2003;11(6):424-7.
9. Sininger Y, Doyle K, Moore J. The case for early identification of hearing loss in children. Pediatr Clin North Am. 1999;46(1):1-14.
10. Oliveira P, Castro F, Ribeiro A. Surdez infantil.Rev Bras Otorrinolaringol.2002;68(3):417-23.
11. Joint Committee on Infant Hearing- JCIH. Position Statement-2000. Am Acad Audiol. 2000;9:1-40.
12. Lima GML, Colella-Santos MF. Triagem auditiva neonatal. In: Marba STM, Mezzacappa Filho F. Manual de Neonatologia Unicamp, 2.ed, Rio de Janeiro: Revinter; 2009. p.408-11.
13. Durieux-Smith A, Fitzpatrick E, Whittingham J. Universal newborn hearing screening: a question of evidence. Int J Audiol. 2008,47(1):1-10.
14. Hyde ML. Newborn hearing screening programs: Overview. J Otolaryngol. 2005;34(Suppl 2):S70-8.
15. Gravel JS, Hood LJ. Avaliação audiológica infantil. In: Musiek FE, Rintelmann WF. Perspectivas atuais em avaliação auditiva. São Paulo: Manole; 2001. p.301-22.
16. Kalatzis V, Petit C. The fundamental and medical impacts of recent progress in research on hereditary hearing loss. Hum Mol Genet. 1998;7(10):1589-97.
17. Declau F, Boudewyns A, van den Ende J, Peeters A, Heyning P. Analysis of 170 referred neonates etiologic and audiologic evaluations after universal neonatal hearing screening. Pediatrics. 2008;121(6):1119-26.
18. Joint Committee on Infant Hearing. Year 2007 Position statement: Principles and guidelines for early hearing detection and intervention programs. Pediatrics. 2007;120(4):898-921.
19. Casali RL, Colella-Santos MF. Auditory Brainstem Evoked Response: response patterns of full-term and premature infants.Braz J Otorhinolaryngol.2010;76(6):729-38.
20. Linares AE, Carvallo RMM. Medidas imitanciométricas em crianças com ausência de emissões otoacústicas. Braz J Otorhinolaryngol. 2008;74(3):410-6.
21. Jerger J. Clinical experience with impedance audiometry. Arch Otolaryngol.1970;92(4):311-23.
22. Carvallo RMM. Fonoaudiologia: Informação para formação. São Paulo: Guanabara-Koogan; 2003. p.344-52.
23. Redondo MC, Lopes Filho O. Testes básicos da avaliação auditiva. In: Lopes Filho O. Tratado de Fonoaudiologia. 2ed. Ribeirão Preto: Tecmedd; 2005. p.89-110.
24. Silman S, Silvermam CA. Basic Audiologic testing. In: Silman S, Silverman CA. Auditory diagnosis: principles and applications. San Diego: Singular Publishing Group; 1997. p.44-52.
25. del Castillo FJ, Rodríguez-Ballesteros M, Alvarez A, Hutchin T, Leonardi E, de Oliveira CA, et al. A novel deletion involving the connexin 30 gene del (GJB6-D13S1854) found in trans with mutations in the GJB2 gene (connexin 26) in subjects with DFNB1 nonsyndromic hearing impairment. J Med Genet. 2005;42(7):588-94.
26. Northern JL, Downs MP. Audição na Infância. 5ªed. Rio de Janeiro: Guanabara Koogan; 2005.
27. Comitê Multidisciplinar em saúde Auditiva - COMUSA. Saúde auditiva neonatal e triagem auditiva neonatal universal-TANU. http://www.audiologiabrasil.org.br/pdf/COMUSA_final_17_maio2009.
28. Cone-Wasson B, Vohr BR, Sininger Y, Widen JE, Folsom RC, Gorga MP, et al. Identification of Neonatal Hearing Impairment: infants with Hearing loss. Ear Hear. 2000;21(5):488-507.
29. Boone RT, Bower CM, Martin PF. Failed newborn hearing screens as presentation for otitis media with effusion in the newborn population. Int J Pediatr Otorhinolaryngol. 2005;69(3):393-7.
30. Saes SO, Goldenberg TBL, Montovani JC. Secreção na orelha média em lactentes- ocorrência, recorrência e aspectos relacionados. J Pediatr (Rio J). 2005;81(2):133-8.
31. Almeida CIR, Almeida RR. Orelha média aguda. In: Lopes Filho O, Campos CAH. Tratado de Otorrinolaringologia. São Paulo: Roca; 2004.
32. Harlor ADB, Bower C. Committee on Practice and Ambulatory Medicine and the Section on Otolaryngology Head and Neck Surgery. Pediatrics. 2009;124:1252-63.
33. Pereira PKS, Azevedo MF, Testa JR. Conductive impairment in newborn who failed the newborn hearing screening. Braz J Otorhinolaryngol 2010;76(6):347-54.
34. Weckx LLM. Tratamento preventivo das otites média. In: Lavinsky L. Tratamento em Otologia. Rio de Janeiro: Revinter; 2006.
35. Doyle KJ, Kong YY, Strobel K, Dallaire P, Ray RM. Neonatal middle ear effusion predict chronic otitis media with effusion. Otol Neurotol. 2004;25(3):318-22.
36. Thomé DC, Sanchez TG, Bento RF. Síndrome de Down e o otorrinolaringologista: características gerais e aspectos otológicos (parte I). Arq Int Otorrinolaringol. 1999;3(30):93-8.
37. Strome M. Down's Syndrome: A modern otorhinolaryngologic perspective. Laryngoscope. 1981;91(10):1581-94.
38. Sininger Y, Hood L, Starr A, Berlin C, Pinton T. Hearing loss due to auditory neuropathy. Audiol Today. 1995;7:10-3.
39. Rapin I, Gravel J. Auditory neuropathy: physiologic and pathologic evidence calls for more diagnostic specificity. Int J Pediatr Otorhinolaryngol. 2003;67(7):707-28.
40. Sanyelbhaa Talaat H, Kabel AH, Samy H, Elbadry M. Prevalence of auditory neuropathy (AN) among infants and young children with severe to profound hearing loss. Int J Pediatr Otorhinolaryngol. 2009;73(7):937-9.
41. Foerst A, Beutner D, Lang-Roth R, Huttenbrink KB, von Wedel H, Walger M. Prevalence of auditory neuropathy/dyssynchrony in a population of children with profound hearing loss. Int J Pediatr Otorhinolaryngol. 2006;70(8):1415-22.
42. Sartorato EL, Gottardi E, Oliveira CA, Magna LA, Annichino-Bizzachi JM, Seixas CA, et al.Determination of carrier frequency of the 35delG mutation in Brazilian neonates. Clin Genet. 2000;58(4):339-40.
43. Tekin M, Arnos KS, Pandya A. Advances in hereditary deafness. Lancet. 2001;358(9287):1082-90.
1. Doutor, Professor Doutor e Coordenadora do Curso de Fonoaudiologia da Faculdade de Ciências Médicas da Unicamp.
2. Doutor, Professor Doutor do Curso de Fonoaudiologia da Faculdade de Ciências Médicas da Unicamp.
3. Doutor, Professor Doutor do Curso de Fonoaudiologia da Faculdade de Ciências Médicas da Unicamp.
4. Doutor, Professor Doutor do Curso de Fonoaudiologia da Faculdade de Ciências Médicas da Unicamp.
5. Mestre, Fonoaudióloga do Setor de Neonatologia do Hospital da Mulher Prof. Dr. José Aristodemo Pinotti - CAISM.
6. Doutor, Médico Otorrinolaringologista do Departamento de Oftalmo/otorrinolaringologia da Faculdade de Ciências Médicas da Unicamp.
7. Doutor, Pesquisadora do Centro de Biologia Molecular e Genética da Unicamp.
Universidade Estadual de Campinas - UNICAMP.
Endereço para correspondência:
Rua Tessalia Vieira de Camargo, 126
Campinas - SP. CEP: 13083-970.
Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) da BJORL em 19 de março de 2011. cod. 7654
Artigo aceito em 17 de junho de 2011.
CNPq e FAEPEX/Unicamp.
Imprimir: ![]()