

Ano: 2011 Vol. 77 Ed. 6 - Novembro - Dezembro - (14º)
Seção: Artigo Original
Páginas: 768 a 774
 PDF PT
PDF PT Achados audiológicos e eletrofisiológicos de indivíduos com a síndrome G/BBB
Auditory findings and electrophysiologics in individuals with G/BBB syndrome
Autor(es): Tatiana Vialôgo Cassab1; Sthella Zanchetta2; Célia Maria Giacheti3; Neivo Luiz Zorzetto4; Antonio Richieri Costa5
Palavras-chave: audição, hipertelorismo, hipospádia, potenciais evocados auditivos do tronco encefálico.
Keywords: evoked potentials, auditory, brainstem, hearing, hypertelorism, hypospadias.
Resumo:
A síndrome G/BBB é uma condição rara, caracterizada por hipertelorismo, fissura de lábio e palato e hipospádia. Não foram encontrados trabalhos sobre a audição em indivíduos com esta síndrome.Objetivo: Investigar a função auditiva em pacientes com síndrome G/BBB quanto à ocorrência ou não de perda auditiva e a condução nervosa auditiva periférica e central. Material e Método: Catorze pacientes de 7 a 34 anos, do gênero masculino, com a síndrome G/BBB, foram avaliados por meio de otoscopia, audiometria, timpanometria e potenciais evocados auditivos de tronco encefálico (PEATE). Forma de Estudo: Estudo de série clínico prospectivo. Resultados: Limiares audiométricos normais em 12 (66,7%) pacientes da amostra e alterados em dois (33,3%), sendo um com perda condutiva e um neurossensorial. Quanto ao PEATE, foram encontrados: latências absolutas da onda I normais em todos os pacientes, aumento das latências absolutas da onda III e V em dois e seis pacientes respectivamente; latências interpicos I-III, III-V e I-V aumentadas em quatro, três e oito pacientes, respectivamente. Conclusão: Perdas auditivas periféricas podem ocorrer na síndrome G/BBB. Há evidências de comprometimento das vias auditivas centrais em nível do tronco encefálico. Estudos com exames de imagem são necessários para maior clareza dos achados clínicos.
Abstract:
The G/BBB syndrome is a rare condition characterized by hypertelorism, cleft lip and palate, and hypospadias. No studies were found on the hearing of individuals with this syndrome. Aim: To investigate the auditory function in patients with G/BBB syndrome, such as the occurrence of hearing loss, and central and peripheral auditory nerve conduction. Methods: Fourteen male patients aged 7-34 years with the G/BBB syndrome were assessed by otoscopy, audiometry, tympanometry and evoked auditory brainstem response (ABR). Model: A retrospective clinical series study. Results: Audiometric thresholds were normal in 12 (66.7%) of the sample and altered in two (33.3%), one with conductive and one with sensorineural loss. ABR resuts were: all patients had normal absolute wave I latencies; absolute wave III and V latencies were increased in two and six patients, respectively; interpeak latencies I-III, IV and V interpeak latencies were increased in four, three and eight patients respectively. Conclusion: Hearing loss can occur in the G/BBB syndrome. There is evidence of central auditory pathway involvement in the brainstem. Imaging studies are needed to clarify the clinical findings.
![]()
INTRODUÇÃO
A síndrome G/BBB foi inicialmente descrita em 19691 e subdividida em duas outras síndromes distintas posteriormente. A síndrome G, assim designada por ter sido descrita em uma família cujo nome tinha por inicial a letra G e que possuía quatro irmãos acometidos, e a síndrome BBB, descrita em três famílias diferentes, todas com nomes iniciados pela letra B, que possuíam oito homens acometidos. Posteriormente, chegou-se à conclusão de que as síndromes G e BBB faziam parte da mesma condição, que apresenta como características defeitos no plano mediano do corpo, sobretudo no esqueleto craniofacial e no tubérculo genital. Seus principais elementos de diagnóstico clínicos são a presença de telecanto e de hipospádia no sexo masculino; e, no feminino, de telecanto e acometimento de hipospádia em parentes do sexo masculino. Sua prevalência é indeterminada, porém, há acometimento de 37 homens para cada 31 mulheres, conforme os casos já relatados2. Não foram encontrados dados sobre prevalência em diferentes raças.
Ainda não se conhece sua forma de transmissão genética, mas a herança deve ser autossômica dominante ou ligada ao X causada pela mutação específica no gene MID1. Recentemente, estudos genéticos moleculares têm demonstrado que ela representa uma condição heterogênea localizada no cromossomo 22 na região q 11.23-12 e no cromossomo X na região p 229,10.
A literatura é ampla no que se refere à associação entre anomalias craniofaciais e perda auditiva. Estudo13 descreve que, de 71 síndromes e anomalias congênitas associadas com surdez, 58 apresentaram algum tipo de anomalia craniofacial. As anomalias craniofaciais congênitas podem estar associadas com distúrbios de audição, especialmente quando a fissura palatina faz parte do quadro clínico. Entre a grande diversidade de síndromes craniofaciais descritas, a síndrome G/BBB é uma condição rara, caracterizada por três principais anomalias: hipertelorismo, fissura de lábio e palato e hipospádia; embora possa haver outras anomalias associadas. A fissura labiopalatina é uma malformação congênita que ocupa uma posição de destaque entre aquelas que cursam com uma complexidade de alterações, especialmente as que se referem aos problemas otológicos e consequente perda de audição.Essas alterações presentes na população com fissura palatina estão relacionadas ao mau funcionamento da tuba auditiva, decorrente da incompetência do músculo tensor do véu palatino, resultando em uma obstrução funcional da tuba e pressão negativa da orelha média, ocasionando otite média e/ou perdas de audição14-18.
A grande ocorrência de perda auditiva nesta população e frequentes histórias de otite média podem interferir no feedback auditivo, privando os indivíduos de experiências verbais e convívio social pleno. Essa privação sensorial pode acarretar prejuízo no desenvolvimento da fala, da linguagem e da aprendizagem18.
Tendo em vista o número de indivíduos regularmente matriculados no Hospital de Reabilitação de Anomalias Craniofaciais-USP com o diagnóstico genético clínico da síndrome G/BBB e a inexistência de estudos relacionados à audição na literatura nacional e internacional, faz-se necessária a investigação sistemática desta população.
A síndrome G/BBB é uma malformação congênita caracterizada por alterações de linha média, como hipertelorismo, hipospádia, fissura de lábio e palato e anomalias laríngeas. Outros sinais que podem ser encontrados nos indivíduos com esta síndrome são ânus imperfurado, atraso no desenvolvimento e defeitos cardíacos19. Entre os principais sinais clínicos presentes na síndrome G/BBB, estão as anomalias estruturais do sistema nervoso central, entre eles, a agenesia ou hipoplasia de corpo caloso, anomalia de Dandy-Walker, alargamento da cisterna magna, alargamento do quarto ventrículo, alargamento da cisterna cerebelar superior, atrofia do vermis, dilatação ventricular, atrofia cerebral e assimetria hemisférica19-22.
De acordo com a literatura compilada, são inexistentes os trabalhos sobre audição periférica ou central na síndrome G/BBB. Existem poucas informações sobre as manifestações comportamentais de indivíduos com a síndrome G/BBB. Sendo assim, o objetivo deste trabalho foi investigar a função auditiva, periférica e central, em pacientes com o diagnóstico da síndrome G/BBB, quanto à ocorrência ou não de perda auditiva, e a condução nervosa auditiva periférica e central, em nível do tronco encefálico.
MATERIAL E MÉTODO
O trabalho foi iniciado após aprovação do Comitê de Ética em Pesquisa da Instituição, atendendo aos dispositivos das Resoluções 196/96 e 251/97 e aprovado sob o protocolo nº 376/2006-UEP-CEP.
Foram avaliados 14 pacientes na faixa etária de 7 a 34 anos, do gênero masculino, com o diagnóstico genético clínico da Síndrome G/BBB. Todos os pacientes apresentavam fissura labiopalatina operada. A bateria de testes audiológicos foi composta por audiometria tonal e vocal, timpanometria e PEATE.
Para esta pesquisa, foi realizado o registro ipsilateral, o estímulo apresentado foi o click, com duração de 0,1 ms, com a frequência de apresentações de 24 clicks por segundo, com espectro de frequência entre 1 e 4 kHz, cuja faixa principal encontrava-se entre 2, 3 e 4 kHz. O tempo de análise utilizado foi de 10ms e, quando necessário, aplicado o filtro, com corte para frequências abaixo de 300 Hz. As ondas foram registradas com o somatório de 1000 estímulos, com polaridade alternada e a intensidade pesquisada foi de 80 dB, sendo efetuados três registros para cada intensidade. O estímulo foi apresentado por meio de fones de ouvido do tipo supra-aurais, modelo Beyerdynamic DT48, marca HORTMANN Neuro-Otometrie.
RESULTADOS
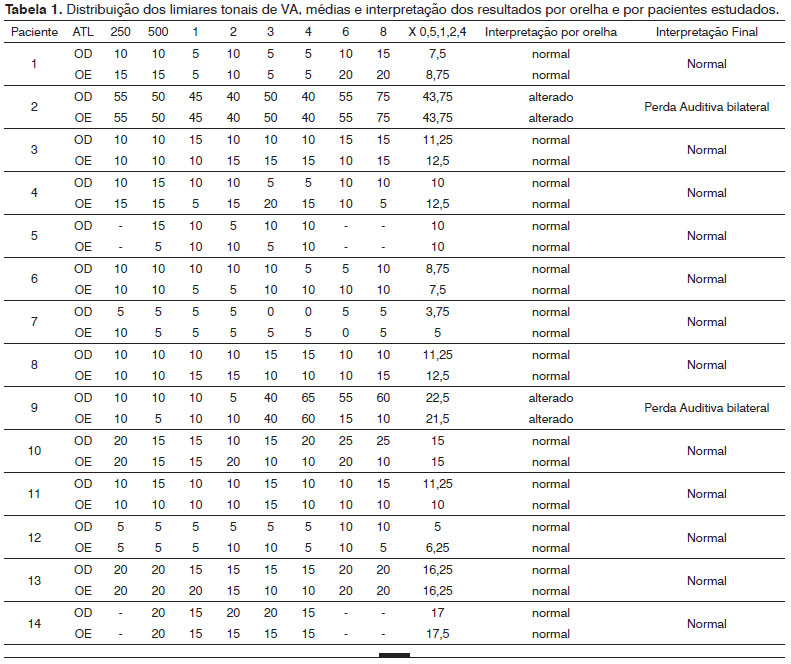
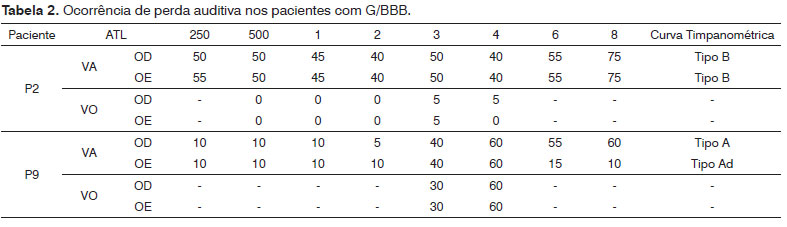
Conforme pode ser constatado, 14,3% (2/14) apresentaram perda auditiva, bilateral. Em relação ao grau da perda, o paciente 2 apresentou perda auditiva de grau moderado, com médias de 43,7 dBNA bilateralmente. O paciente 9 apresentou perda auditiva de grau leve, com média de 22.5 dBNA e 21.25 dBNA para as orelhas direita e esquerda, respectivamente.
Dos 14 pacientes da amostra, a timpanometria foi realizada em 13, pelo fato de um paciente utilizar tubos de ventilação bilateralmente. Verificamos, assim, que, das 26 orelhas testadas, a maioria (19 - 73%) apresentou curva tipo A, revelando integridade da orelha média. Foi obtida curva tipo Ad em três orelhas (11,5%), tipo B em duas orelhas (7,7%) e tipo As em duas (7,7%) das orelhas testadas (Tabela 1).
Conforme pode ser constatado, 14,3% (2/14) apresentaram perda auditiva, bilateral. Em relação ao grau da perda, o paciente 2 apresentou perda auditiva de grau moderada, com médias de 43,7 dBNA bilateralmente. O paciente 9 apresentou perda auditiva de grau leve, com média de 22.5 dBNA e 21.25 dBNA para as orelhas direita e esquerda, respectivamente (Tabela 2).
Dos 14 pacientes da amostra, a timpanometria foi realizada em 13, pelo fato de um paciente utilizar tubos de ventilação bilateralmente.
Verificamos, assim, que, das 26 orelhas testadas, a maioria 19 (73%) apresentou curva tipo A, revelando integridade da orelha média. Foi obtida curva tipo Ad em três orelhas (11,5%), tipo B em duas orelhas (7,7%) e tipo As em duas (7,7%) das orelhas testadas.
Dentre os 14 pacientes da amostra, os dois com perda auditiva periférica foram excluídos. Assim sendo, a população estudada neste capítulo foi composta por 12 indivíduos.
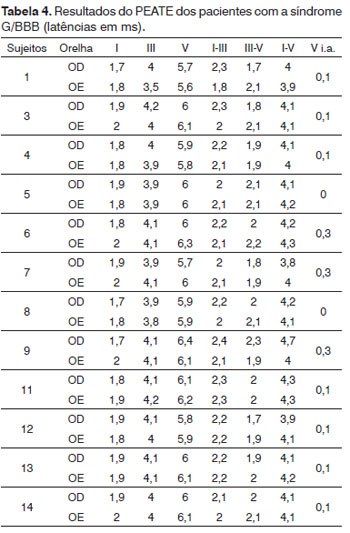
Na Tabela 3, podemos observar os valores médios dos parâmetros LA e LIP, obtidos nos pacientes.
Todos os 12 pacientes apresentaram as ondas I, III e V bilateralmente e com boa morfologia. Em relação ao parâmetro de latência, quatro pacientes apresentaram valores dentro do padrão de referência, o que corresponde a 33,3% da amostra. Os outros oito pacientes (66,7%) apresentaram valores superiores aos adotados e foram considerados como alterados, sendo que em seis deles a alteração foi bilateral e, em dois, unilateral. Nenhum deles apresentou diferença interaural da onda V maior que 0,4ms.
Os resultados individuais das LA e LIP do PEATE de toda a casuística encontram-se na Tabela 4.
As latências absolutas da onda I foram encontradas dentro dos padrões de normalidade em todos os pacientes avaliados.
Quanto aos valores das latências absolutas, observou-se que dois pacientes apresentaram aumento da latência da onda III unilateralmente e quatro apresentaram aumento da latência onda V, sendo um bilateral e três unilateralmente.
Para os valores das latências interpicos, foi observado que quatro pacientes apresentaram aumento do interpico I-III, sendo um bilateral e três unilateralmente; dois do interpico III-V unilateralmente e seis do interpico I-V, sendo dois bilateral e quatro unilateralmente.
DISCUSSÃO
Não foi possível a comparação direta dos achados com a literatura compilada, uma vez que não foram encontrados trabalhos com o objetivo de estudar a audição na síndrome G/BBB. Analisando os dados obtidos na história clínica, podemos observar que quatro dos 14 pacientes entrevistados relataram suspeita de hipoacusia e histórico de otites de repetição, dos quais dois apresentaram alterações audiométricas. A ocorrência de otite média em indivíduos com fissura de palato é bem documentada na literatura14,15, podendo gerar ou não queixas auditivas, tendo em vista que o quadro de otite média serosa pode apresentar poucos sintomas. Alguns autores23 observaram que grande parte dos indivíduos com fissura palatina não operada e perda auditiva não apresentaram queixas auditivas quando questionados. Apesar de todos os pacientes avaliados no presente estudo apresentarem fissura de palato como parte de seus sinais clínicos, a maioria da amostra não apresentou queixa auditiva, e apenas dois apresentaram perda auditiva periférica.
A audiometria tonal foi realizada em todos os pacientes da amostra e os resultados foram analisados quantitativa e qualitativamente.
Com relação aos achados quantitativos, pode-se verificar que dois dos 14 pacientes avaliados apresentaram perda auditiva com comprometimento bilateral, sendo um do tipo neurossensorial de grau leve e um do tipo condutivo e grau moderado, ambos compatíveis com a timpanometria, que revelou normalidade de orelha média no primeiro e alteração no segundo, respectivamente, mostrando, também, concordância com os dados da entrevista audiológica. Da mesma forma, a presença de perda auditiva neurossensorial no paciente supracitado pode ser justificada pela alteração condutiva com frequentes histórias de recidivas23-25.
Apesar de a população com fissura labiopalatina ser considerada bastante vulnerável em relação à saúde auditiva, devido ao funcionamento inadequado da tuba auditiva, em virtude da inserção anormal dos músculos elevador e tensor do véu palatino4,8, os pacientes estudados encontraram-se com audição dentro dos padrões de normalidade, mostrando concordância com os achados timpanométricos.
A casuística estudada encontrou-se em faixa etária de 7 a 34 anos e todos já haviam realizado palatoplastia previamente. Estes fatores provavelmente contribuíram para a baixa ocorrência de alterações condutivas no presente estudo, uma vez que a melhora significativa da audição após a palatoplastia, relacionada ao restabelecimento anatomofuncional da musculatura do véu palatino, é bem documentada na literatura26-30. Além disso, tanto na população com fissura palatina como em indivíduos sem esta malformação, as alterações de orelha média são mais comuns na primeira infância, devido à posição mais horizontalizada da tuba auditiva nesta faixa etária31.
Para a análise dos resultados do PEATE, foram usados os valores do padrão de normalidade obtidos mediante calibração biológica32 utilizados no setor de genética clínica do Hospital de Reabilitação de Anomalias Craniofaciais (HRAC).
Analisando os resultados, pode-se verificar que não houve nenhum paciente com ausência das principais ondas analisadas; entretanto, a maior parte da amostra (66,7%) apresentou latências absolutas e interpicos aumentadas.
Observou-se neste estudo que as latências absolutas da onda III (gerada no núcleo coclear e complexo olivar) e da onda V (gerada no lemnisco lateral e complexo olivar inferior), bem como os interpicos I-III, III-V e I-V, encontraram-se aumentadas em oito dos 12 pacientes avaliados por este procedimento quando comparados ao padrão de normalidade.
Sendo assim, os resultados descritos sugerem que pacientes com síndrome G/BBB apresentam um atraso na condução do estímulo acústico ao longo da via auditiva. Alterações na condução neural do estímulo sonoro com limiares audiométricos normais foram descritas na literatura33. Estes autores verificaram aumento nas latências das ondas III e V e dos interpicos I-III e IV do PEATE em indivíduos com audição dentro dos padrões de normalidade.
Alterações do tipo condutivo causam aumento de latências absolutas no PEATE30,34,35, o que justifica a não realização do PEATE nos dois pacientes do estudo que apresentaram alterações auditivas periféricas.
As alterações de PEATE encontradas no presente estudo também podem ser justificadas pelo possível comprometimento do sistema nervoso central, pois a literatura relata que, entre os principais sinais clínicos presentes na síndrome G/BBB, estão as anomalias estruturais do sistema nervoso central19-22. Apesar disso, não foi possível a confirmação do comprometimento do SNC nos pacientes avaliados, devido a dificuldades técnicas para a realização de exames por imagem, constituindo em limitação do presente estudo. Estudos futuros, correlacionando os achados de imagem com os achados audiológicos, poderão ajudar a responder esta questão.
Anomalias craniofaciais associadas a síndromes genéticas são consideradas fatores de risco para alterações auditivas36. No presente estudo, a ocorrência da fissura labiopalatina em todos os pacientes estudados pode ser considerada um viés de amostra, visto que o local onde o estudo foi desenvolvido é um hospital especializado no tratamento de anomalias craniofaciais e fissura labiopalatina. O presente estudo reforça a importância da investigação do sistema auditivo em pacientes com a síndromeG/BBB, tendo em vista o importante papel do funcionamento adequado das vias auditivas periféricas e centrais para o desenvolvimento da linguagem e interação social.
CONCLUSÃO
Frente aos resultados obtidos, podemos concluir que pacientes com a síndrome G/BBB podem apresentar perdas auditivas periféricas, condutivas e neurossensoriais. Entretanto, não há subsídios para afirmar que as mesmas são em decorrência da síndrome ou da associação com a fissura de palato.
Há evidências de comprometimento das vias auditivas centrais em nível do tronco encefálico, embora as alterações estruturais do SNC relatadas nesta síndrome não estejam relacionadas diretamente com as vias auditivas.
Estudos com enfoque no perfil audiológico desta população, com exames de imagem, são necessários para maior clareza dos achados clínicos.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Opitz JM, Frias JL, Gutenberger JE, Pellett JR. The G syndrome of multiple congenital anomalies. Birth Defects Orig Artic Ser. 1969;5(2):95-101.
2. Buyse ML. Hypertelorism-Hypospadias Syndrome. In: Buyse ML ed., Birth defects Encyclopedia. Dover: Center for Birth Defects Information Services, Inc.; 1990. p.912-4.
3. Kimmelman CP, Denneny JC. Opitz (G) syndrome. Int J Pediatr Otorhinolaryngol. 1982;4(4):343-7.
4. Cappa M, Borrelli P, Marini R, Neri G. The Opitz syndrome: a new designation for the clinically indistinguishable BBB and G syndromes. Am J Med Genet. 1987;28(2):303-9.
5. Allanson JE. G syndrome: an unusual family. Am J Med Genet. 1988;31(3):637-42.
6. Christodoulou J, Bankier A, Loughan P. Ring chromosome 22 karyotype in a patient with Optiz (BBBG) syndrome. Am J Med Genet. 1990;37(3):422-4.
7. Schrander J, Schrander-Stumpel C, Berg J, Frias JL. Opitz BBBG syndrome: new family with late-onset, serious complication. Clin Genet. 1995;48(2):76-9.
8. Opitz JM. Personal Communication. Helena, Mont., 2/25/1996.
9. Lacassie Y, Arriaza MI. Opitz GBBB syndrome and the 22q11.2 deletion. (Letter). Am J Med Genet. 1996;62(3):318.
10. McDonald-Mcginn DM, Driscroll DA, Bason, L, Christensen K, Lynch D, Sullivan K, et al. Autosomal dominant «OPTIZ» GBBB syndrome due to a 22q11.2 deletion. Am J Med Genet. 1995;59(1):103- 13.
11. So J, Suckow V, Kijas Z, Kalscheuer V, Moser B, Winter J, et al. Mild phenotypes in a series of patients with Opitz GBBB syndrome with MID1 mutations. Am J Med Genet. 2005;132A(1):1-7.
12. Cho HJ, Shin MY, Ahn KM, Lee SI, Kim HJ et al. X-linked Opitz G/BBB syndrome: identification of a novel mutation and prenatal diagnosis in a Korean family. J Korean Med Sci. 2006;21(5):790-3.
13. Jones KL. Smith's recognizable patterns of human malformation. California: Elsevier; 2006.
14. Rood SR, Stool SE. Current concepts of the etiology, diagnosis, and management if cleft palate related otopathologic disease. Otolaryngol Clin North Am. 1981;14(4):865-84.
15. Gravel JS, Wallace IF, Ruben RJ. Early otitis media and latter educational risk. Acta Otolaryngol. 1995;115(2):279-81.
16. Tunçbilek G, Ozgur F, Belgin E. Audiologic and tympanometric findings in children with cleft lip and palate. Cleft Palate Craniofac J. 2003;40(3):304-9.
17. Arnold WH, Nohadani N, Koch KHH. Morphology of the auditory tube and palatal muscles in a case of bilateral cleft palate. Cleft Palate Craniofacial J. 2005;42(2):197-201.
18. Flynn T, Möller C, Jönsson R, Lohmander A. The high prevalence of otitis media with effusion in children with cleft lip and palate as compared to children without clefts. Int J Pediatr Otorhinolaryngol. 2009;73(10):1441-6.
19. Neri G, Genuardi M, Natoli G, Costa P, Maggioni G. A girl with G syndrome and agenesis of the corpus callosum. Am. J. Med. Genet. 1987;28(2):287-91.
20. Williams CA, Frias JL. Apparent G syndrome presenting as neck and upper limb dystonia and severe gastroesophageal reflux. Am J Med Genet. 1987;28(2):297-302.
21. Young ID, Dalgleish R, MacKay EH, MacFadyen UM. Discordant expression of the G syndrome in monozygotic twins. Am J Med Genet. 1988;29(4):863-9.
22. Guion-Almeida ML, Richieri-Costa A. CNS midline anomalies in the Opitz G/BBB síndrome: report on 12 brazilian patients. Am J Med Genet. 1992;43(6):918-28.
23. Ramana YV, Nanda V, Biswas G, Chittoria R, Ghosh S, Sharma RK. Audiological profile in older children and adolescents with unrepaired cleft palate. Cleft Palate Craniofac J. 2005;42(5):570-3.
24. Zambonato TCF, Feniman MR, Blasca WQ, Lauris JRP, Maximino LP. Perfil de usuários de AASI com fissura labiopalatina. Braz J Otorhinolaryngol. 2009;75(6):888-92.
25. Yoshida H, Miyamoto I, Takahashi H. Is sensorineural hearing loss with chronic otitis media due to infection or aging in older patients? Auris Nasus Larynx. 2009;36(3):269-73.
26. Piazentin SHA. A influência da palatoplastia primária nas alterações do ouvido médio [dissertação]. São Paulo: Pontifícia Universidade Católica de São Paulo; 1989.
27. Paradise JL, Elster BA, Tan L. Evidence in infantis with cleft palate that breast milk protects against otitis media. Pediatrics. 1994;94(6 Pt 1):853-60.
28. Piazentin-Penna SHA. Avaliação audiológica em crianças de 3 a 12 meses de idade com fissura labiopalatina [tese]. Bauru: Hospital de Reabilitação de Anomalias Craniofaciais, Universidade de São Paulo; 2002.
29. Goudy S, Lott D, Canady J, Smith RJ. Conductive hearing loss and otopathology in cleft palate patients. Otolaryngol Head Neck Surg. 2006;134(6):946-8.
30. Ernestino GMRC. Estudo comparativo pré e pós-palatoplastia de indivíduos submetidos à avaliação audiológica. [dissertação]. Bauru: Hospital de Reabilitação de Anomalias Craniofaciais, Universidade de São Paulo; 2007.
1. Mestre, Pós-graduanda, nível doutorado, em Ciências da Reabilitação do Hospital de Reabilitação de Anomalias Craniofaciais, HRAC. da Universidade de São Paulo, Bauru, São Paulo, Brasil.
2. Doutora, Docente do Curso de Fonoaudiologia da Faculdade de Medicina de Ribeirão Preto - FMRP/USP.
3. Livre-Docente, Professora Adjunta de Diagnóstico Fonoaudiológico do Departamento de Fonoaudiologia da Universidade Estadual Paulista-UNESP-Marília; São Paulo, Brasil.
4. Livre-Docente, Professor Titular de Anatomia e Fisiologia dos Órgãos da Audição e da Fala do Departamento de Fonoaudiologia da Universidade Estadual Paulista - UNESP-Marília; São Paulo, Brasil.
5. Livre-Docente, Médico Geneticista do Hospital de Reabilitação de Anomalias Craniofaciais, Universidade de São Paulo, USP, Bauru-SP.
Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) da BJORL em 16 de agosto de 2010. cod. 7270
Artigo aceito em 19 de maio de 2011.

Imprimir: ![]()