

Ano: 2011 Vol. 77 Ed. 4 - Julho - Agosto - (24º)
Seção: Relato de Caso
Páginas: 540 a 540
 PDF PT
PDF PT Membrana laríngea anterior e Síndrome de deleção 22q11
Anterior laryngeal membrane and 22q11 deletion syndrome
Autor(es): Rafael Fabiano Machado Rosa1; Rosana Cardoso Manique Rosa2; Rita Carolina Pozzer Krumenauer3; Marileila Varella-Garcia4; Giorgio Adriano Paskulin5
Palavras-chave: cromossomos humanos par 22, hibridização in situ fluorescente, laringe, síndrome de DiGeorge.
Keywords: chromosomes, human, pair 22, DiGeorge syndrome, in situ hybridization, fluorescence, larynx.
![]()
INTRODUÇÃO
Membranas laríngeas anteriores (MLAs) são anormalidades incomuns, caracterizadas pela presença, ao nascimento, de tecido membranoso na região supraglótica, glótica e/ou subglótica1-3. Elas são responsáveis por cerca de 5% das malformações de laringe1 e, dependendo da sua extensão, podem causar obstrução das vias aéreas levando a sintomas como alteração no choro, estridor, disfonia e disfunção respiratória2,4. Indivíduos afetados frequentemente apresentam anomalias associadas, como defeitos cardíacos congênitos e alterações de palato, fazendo parte muitas vezes de síndromes genéticas conhecidas1-3.
Relatamos aqui o caso de um paciente apresentando membrana laríngea anterior e síndrome de deleção 22q11 (SD22q11), também conhecida como síndrome velocardiofacial ou síndrome de DiGeorge (OMIM 188400/192430)5.
RELATO DE CASO
Paciente de 12 anos e 2 meses, sexo masculino, caucasiano, internou-se inicialmente no hospital para realização de cirurgia de correção de comunicação interatrial do tipo fossa oval. Ele é o primeiro filho de um casal de pais jovens, hígidos e não consanguíneos. A história familiar era negativa para defeitos congênitos ou doenças genéticas. Sua gestação cursou sem intercorrências. O paciente nasceu de parto normal, a termo, apresentação cefálica, pesando 3.430 gr. (P50), medindo 50 cm (P25-50) e com perímetro cefálico de 35 cm (P50-98). Ele se mostrava cianótico, sendo que não chorou ao nascimento. Necessitou de oxigenoterapia, ficando hospitalizado durante os primeiros 15 dias de vida. Foi submetido a uma cirurgia de laringe para retirada de membrana subglótica com laser aos 3 meses de idade, sendo que, neste período, apresentou episódios de hipocalcemia com necessidade de tratamento com gluconato de cálcio.
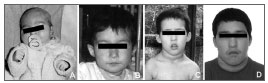
O paciente evoluiu com atraso de desenvolvimento neuropsicomotor e de fala, além de alteração comportamental, com necessidade de tratamento medicamentoso com haloperidol e biperideno. Ao exame físico, aos 12 anos e 2 meses, ele apresentava peso de 47 kg (P75-90), estatura de 147 cm (P50), perímetro cefálico de 53 cm (P2-50), face alongada, fendas palpebrais estreitas, prega epicântica bilateral, hipoplasia das asas do nariz, maloclusões dentárias, prognatismo, sobredobramento dos ramos horizontais e verticais das hélices, pectus carinatum, umbigo protuso, criptorquidia à direita e dedos das mãos afilados. O palato era ogival e não possuía anormalidades associadas, como insuficiência velofaríngea. Na Figura 1 podemos ver as características craniofaciais do paciente em diferentes idades. Como apresentava um quadro de disfonia de causa não conhecida, foi encaminhado à otorrinolaringologia. A avaliação pela nasofibrolaringoscopia identificou a presença de uma MLA associada à estenose subglótica anterior com reduzida luz da região glótica. O aspecto da traqueia e da carina eram normais. A criança não apresentava dispneia, sendo que a conduta foi expectante. A avaliação complementar pelo ultrassom abdominal identificou apenas um baço acessório. Os níveis de cálcio estavam dentro da normalidade. A avaliação citogenética inicial, pelo cariótipo de alta resolução por bandas GTG (> 550 bandas), foi normal (46,XY). A pesquisa de microdeleção 22q11.2 pela técnica de hibridização in situ fluorescente (FISH), utilizando-se da sonda de DNA DiGeorge/VCFS Region Probe (TUPLE 1), evidenciou o diagnóstico de SD22q11.
Figura 1. Aspecto craniofacial do paciente em diferentes idades: aos 16 dias de vida (A), com 1 ano e 11 meses (B), aos 4 anos (C) e aos 13 anos de idade (D).
DISCUSSÃO
A MLA representa a forma mais branda de atresia de laringe (a do tipo III)4. A sua associação com a SD22q11 tem sido descrita na literatura ao longo das últimas duas décadas em poucos trabalhos, sendo a maioria deles formada de relatos com poucos pacientes1-4. Miyamoto et al. (2004)3, em uma das poucas séries de casos, encontraram uma frequência de SD22q11 de 65% entre 17 pacientes com MLA. Por outro lado, estudos avaliando pacientes com a SD22q11 têm observado que esta malformação tem sido descrita em aproximadamente 1 a 2% dos casos6.
A SD22q11 é uma doença genética relativamente comum, causada por uma deficiência na região 11 do braço longo do cromossomo 22. Apesar da maior parte dos casos de SD22q11 ser esporádica (devido a mutações novas), indivíduos com a deleção possuem um risco de 50% de passarem a mesma a seus filhos. A síndrome caracteriza-se clinicamente por um espectro fenotípico bastante amplo que inclui, entre tantas alterações, diversos tipos de alterações otorrinolaringológicas. Isto se reflete nos seus diferentes nomes, dados antes da sua identificação no início da década de 90, que incluem, entre outros, síndrome de DiGeorge, síndrome velocardiofacial e síndrome de Shprintzen. Estes espelham a visão de diferentes especialistas para a mesma doença6.
Contudo, é importante estar ciente que, em alguns casos, especialmente em crianças menores, estas características podem não ser tão evidentes, chamando, assim, pouca atenção quanto à possibilidade diagnóstica da síndrome3,5 (ver Figura 1). A hipocalcemia, tal como em nosso caso, pode ser também latente, tornando-se aparente apenas no momento de um procedimento cirúrgico7.
COMENTÁRIOS
Frente a estes achados, acreditamos, tal como já sugerido na literatura1-4, que pacientes portadores de MLA, em especial aqueles com outras malformações congênitas como defeitos cardíacos, deveriam ser sempre testados para a SD22q11. Este diagnóstico possui importantes implicações dentro do manejo e do aconselhamento genético do paciente e de sua família.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Stoler JM, Ladoulis M, Holmes LB. Anterior laryngeal webs and 22q11 deletions. Am J Med Genet. 1998;79:152.
2. McElhinney DB, Jacobs I, McDonald-McGinn DM, Zackai EH, Goldmuntz E. Chromosomal and cardiovascular anomalies associated with congenital laryngeal web. Int J Pediatr Otorhinolaryngol. 2002;66:23-7.
3. Miyamoto RC, Cotton RT, Rope AF, Hopkin RJ, Cohen AP, Shott SR, et al. Association of anterior glottic webs with velocardiofacial syndrome (chromosome 22q11.2 deletion). Otolaryngol Head Neck Surg. 2004;130:415-7.
4. Fokstuen S, Bottani A, Medeiros PFV, Antonarakis SE, Stoll C, Schinzel A. Laryngeal atresia type III (glotic web) with 22q11.2 microdeletion: report of three patients. Am J Med Genet. 1997;70:130-3.
5. Online Mendelian Inheritance in Man, OMIM (TM) [homepage on the Internet]. Baltimore e Bethesda: BeMcKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University and National Center for Biotechnology Information, National Library of Medicine [cited 2010 Aug 10]. Available from: http://www.ncbi.nlm.nih.gov/omim/.
6. Rosa RFM, Zen PRG, Roman T, Graziadio C, Paskulin GA. Síndrome de deleção 22q11.2: compreendendo o CATCH22. Rev Paul Pediatr. 2009;27:211-20.
7. Schaan BD, Huber J, Leite JC, Kiss A. Cardiac surgery unmasks latent hypoparathyroidism in a child with the 22q11.2 deletion syndrome. J Pediatr Endocrinol Metab. 2006;19:943-6.
1. Mestrado, Médico Geneticista da UFCSPA/CHSCPA e Doutorando do Programa de Pós-Graduação em Patologia da UFCSPA.
2. Especialização em Pediatria, Mestranda pelo Programa de Pós-Graduação em Patologia da UFCSPA, Brasil.
3. Mestrado em Ciências da Saúde, Médica Otorrinolaringologista, Preceptora do Serviço de Otorrinolaringologia do Hospital da Criança Santo Antônio, HCSA./CHSCPA, Brasil.
4. Doutorado em Ciências Biológicas, Professora e Citogeneticista Responsável pelo Laboratório de Citogenética da Division Medical Oncology, University of Colorado Denver, EUA.
5. Doutorado em Genética e Biologia Molecular, Médico Geneticista da UFCSPA/CHSCPA, Professor da Disciplina de Genética Clínica e do Programa de Pós-Graduação em Patologia da UFCSPA, Citogeneticista Responsável pelo Laboratório de Citogenética da UFCSPA, Brasil.
Universidade Federal de Ciências da Saúde de Porto Alegre (UFCSPA), Complexo Hospitalar Santa Casa de Porto Alegre (CHSCPA) e University of Colorado.
Endereço para correspondência:
Prof. Dr. Giorgio Adriano Paskulin
Rua Sarmento Leite, 245/403, Bairro, Centro
Porto Alegre - RS, Brasil
Tel. (0xx51) 3303-8771 - Fax: (0xx51)33038810
Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) da BJORL em 15 de julho de 2010. cod. 7209
Artigo aceito em 1 de outubro de 2010.
University of Colorado (EUA) e Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (Brasil).
Imprimir: ![]()