

Ano: 2008 Vol. 74 Ed. 6 - Novembro - Dezembro - (2º)
Seção: Artigo Original
Páginas: 807 a 814
 PDF PT
PDF PT Dados clínicos e da audição em indivíduos com Síndrome de Alport
Clinical data and hearing of individuals with Alport syndrome
Autor(es): Fatima Regina Abreu Alves1, Fernando de Andrade Quintanilha Ribeiro2
Palavras-chave: colágeno tipo iv, genética, nefrite hereditária, perda auditiva.
Keywords: hereditary, genetics, nephritis, hearing loss.
Resumo:
A Síndrome de Alport (SA) é uma desordem hereditária, caracterizada por nefropatia, muitas vezes, com perda auditiva sensorioneural e com defeitos oculares. Objetivo: Analisar os dados clínicos e da audição em indivíduos com SA, com ênfase na correlação entre alteração renal e perda auditiva (PA). Forma de Estudo: clínico prospectivo com coorte transversal. Casuística e Método: 37 indivíduos realizaram avaliação otorrinolaringológica e testes audiológicos. Foram considerados para a análise estatística da PA os resultados das audiometrias de tons puros. Resultados: Nos 28 indivíduos que apresentavam alterações clínicas foram encontrados 46,4% de DLX e 53,6% de AD. A PA ocorreu em 46,1% dos avaliados. 12 pacientes tinham PA no exame audiométrico 11,5%, leve e 34,6%, moderada. Comparados os familiares normais aos que tinham alteração renal, todos os que apresentavam PA mostravam comprometimento renal. Em 30,8% a configuração era descendente suave em agudos e em 11,5% era plana. Conclusões: A distribuição dos padrões de herança não coincide com o descrito na literatura. A PA é um achado extra-renal freqüente. Existe associação entre acometimento renal e PA (p= 0,009). As configurações mais freqüentes foram: descendente suave em agudos e plana. Não há associação entre a PA e a idade. Não existe correlação entre PA e sexo neste grupo.
Abstract:
Alport Syndrome (AS) is a hereditary disease, characterized by nephropathy, often times with sensorineural hearing loss and ocular defects. Aim: to analyze the clinical and hearing information from individuals with AS, more specifically the correlation between renal disorder and hearing loss (HL). Study design: clinical prospective with cross-sectional cohort. Materials and Methods: 37 individuals underwent otorhinolaryngological evaluation and were submitted to audiologic tests. For HL statistical analysis we considered only the results from the pure tone audiometries. Results: of the 28 individuals with clinical alterations, we found 46.4% of DLX and 53.6% of AD. HL happened to 46.1% of the individuals evaluated. 12 patients presented HL in the audiometric test: 11.5% mild and 34.6% moderate. Comparing the normal relatives with those with renal disorder; all that had HL also had renal disorder. In 30.8% the curve shape was mild descending in the high frequencies and in 11.5% it was flat. Conclusions: The inheritance pattern distribution does not match literature descriptions. HL is a frequent extra-renal finding. There is an association between renal involvement and HL (p= 0.009). The most frequent curve shapes: mild descending in the high frequencies and flat. There was no association between HL and age. There is no correlation between the HL and gender in this group.
![]()
INTRODUÇÃO
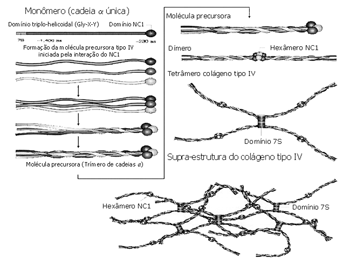
A Síndrome de Alport (SA) é uma desordem hereditária caracterizada por hematúria, que leva freqüentemente à falência renal. Pode ser acompanhada de manifestações extra-renais. Muitas vezes, portanto, a nefropatia está associada com perda auditiva sensorioneural (PASN) e defeitos oculares.1 A forma dominante ligada ao X (DLX) ocorre em 85% dos casos, a forma autossômica recessiva (AR), em 10 a 15% dos casos, e a forma autossômica dominanteé rara.2 O aprimoramento do diagnóstico molecular pode evidenciar que o padrão AD não é tão infreqüente.3 Mutações no gene COL4A5 explicam grande parte e, possivelmente, toda a SAligada ao X.4 Para o entendimento da SA é necessário conhecer a estrutura do colágeno tipo IV (Figura 1).5 O colágeno tipo IV é o principal constituinte das membranas basais. As cadeias a3, a4 e a5 (IV) expressam-se seletivamente nas membranas basais de alguns tecidos, como: o rim (membranas basais glomerulares), a cóclea e o olho. As mutações presentes na SA produzem defeitos nas cadeias a3, a4 e a5 (IV). Estes defeitos resultam no enlaçamento e na montagem incorreta dos monômeros, que serão degradados rapidamente.Os sinaisa seguir são observados no quadro clínico da SA. A hematúria é um sinal cardinal e observada no primeiro ano de vida. A proteinúria reflete comprometimento glomerular específico, estando ausente nos primeiros anos de vida. A falência renal é progressiva. A PASN é de intensidade variável, bilateral, simétrica e progressiva. Ocorre, mais freqüentemente, em homens do que em mulheres na forma DLX. Alterações oculares: o lentecone anterior (ocorre em 10 a 30% dos casos, é patognomônico da SA), manchas oculares, catarata, nistagmo e miopia. A hematúria é precoce, a PASN evolui durante os anos escolares, oestágio final de falência renal e alterações oculares ocorre no fim da aolescência.6
Figura 1. Estrutura do colágeno tipo IV. - As cadeias x3, x4 e x5(IV) expressam-se nas membranas basais do rim, da cóclea e do olho. As mutações presentes na SA produzem defeitos nestas cadeias. O dano ao colágeno tipo IV, devido à mutação, rompe a função de ligação epitelial e leva ao defeito do órgão. Modificado de Kalluri,2003. Gly=glicina; X=prolina; Y=hidroxiprolina; NC1= porção globular carboxi-terminal não colagenosa; 7S= domínio amino-terminal; aa=aminoácidos.
OBJETIVOS
O objetivo é analisar os dados clínicos e da audição, em indivíduos com Síndrome de Alport, com ênfase na correlação entre alteração renal e perda auditiva.
CASUÍSTICA E MÉTODO
O Projeto de Pesquisa e o Termo de Consentimento Livre e Esclarecido foram aprovados pelo Comitê de Ética em Pesquisa daInstituição (Projeto nº 118/05). Realizado um estudo clínico prospectivo comcoorte transversal.
Foram avaliadas oito famílias e elaborados os respectivos heredogramas, com informações de pelo menos mais uma geração. Os dados clínicos e audiológicos dos casos índices (probandos; membros a partir dos quais uma família comdistúrbio genético é inicialmente avaliada) encaminhados foram colhidos e realizaram-se avaliações nos membros das famílias, que concordaram em participar. Todas as famílias preenchiam pelo menos dois dos critérios diagnósticos estritos.
Trinta e sete indivíduos destas famílias aceitaram participar do estudo e realizaram uma avaliação composta de: histórias clínica e familiar detalhadas, exame físico otorrinolaringológico completo, avaliação de resultados prévios de exames laboratoriais (de sangue, de urina, de ultra-sonografia e de biópsia renal). As famílias dois, cinco, seis e oito tinham indivíduos submetidos à biópsia renal, com alterações histológicas compatíveis com a Síndrome de Alport. Os pacientes não apresentavam história de exposição a ruído e nem uso de ototóxicos. As famílias um, quatro e cinco apresentavam indivíduos com alterações oculares. Não foi observado o lentecone anterior.
Os testes audiológicos foram realizados seguindo a mesma padronização, nos casos índices e em outros membros da família que aceitaram participar do projeto. Crianças abaixo de três anos de idade que não responderam adequadamente aos testes ou não puderam ser condicionadas, para fins estatísticos, foram consideradas como não tendo feito os exames audiométricos.
Os resultados foram classificados, considerando-se a média em 500, 1.000, 2.000 e 4.000 Hz, do pior ouvido, em: normal; leve (21 - 40 dB NA); moderado (41 - 60 dB NA); moderadamente severo (61 - 80 dB NA); severo (81 - 100 dB NA); profundo (> 100 dB NA) e não-realizado.7
Uma diferença de 15 dB entre as orelhas em pelo menos duas freqüências ou uma diferença de 10 dB em quatro freqüências define a perda auditiva assimétrica e a perda simétrica apresenta a mesma configuração e diferença < 15 dB nas mesmas freqüências entre as orelhas.7
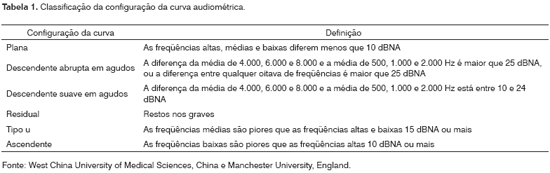
As curvas audiométricas foram classificadas, conforme o descrito na Tabela 1, em planas, descendente abrupta em agudos, descendente suave em agudos, específicas (tipo U ou ascendentes) e residual.8
Os heredogramas das oito famílias foram elaborados com os dados coletados e com os resultados dos exames audiológicos, destacando-se os que participaram do estudo (em vermelho) daqueles em que apenas foram obtidas informações.
Utilizando-se o software EpiInfo, versão 3.2, foram aplicados os seguintes testes estatísticos: Qui-quadrado (c2) e Teste Exato de Fisher. Foi considerado um intervalo de confiança de 95% nas comparações estatísticas.
RESULTADOS
1 - Heredogramas
Usando símbolos padrão, as informações obtidas sobre a história familiar dos probandos avaliados foram representadas graficamente nos heredogramas a seguir (Figuras 2-9).
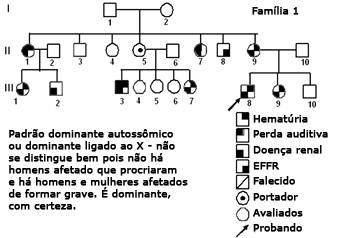
Figura 2. Heredograma família 1 - Provavelmente autossômico dominante, com expressividade variável. Tem homens e mulheres igualmente comprometidos.
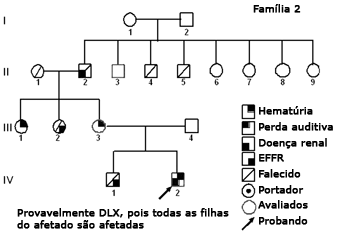
Figura 3. Heredograma família 2 - Provavelmente DLX, pois todas as filhas do afetado II-2 são afetadas.
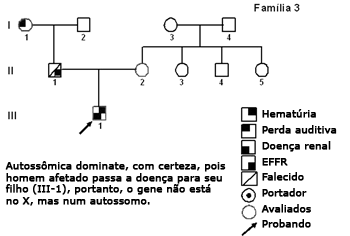
Figura 4. Heredograma família 3 - Autossômica dominante, pois homem afetado (II-1) passa a doença para seu filho (III-1), portanto o gene não está no X, mas num autossomo.
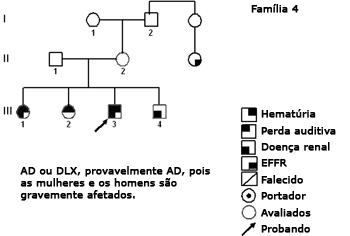
Figura 5. Heredograma família 4 - Provavelmente AD. Os casos III-1, III-3 e III-4 estão bem comprometidos. As mulheres DLX têm um acometimento mais brando que os homens.
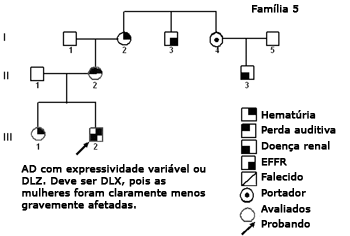
Figura 6. Heredograma família 5 - Provavelmente DLX. O contraste na severidade entre homens e mulheres é compatível com este padrão de herança.
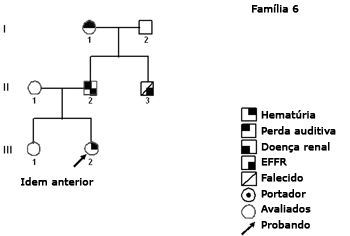
Figura 7. Heredograma família 6 - Provavelmente DLX. A característica de um heredograma DLX, totalmente penetrante, é que todas as filhas e nenhum dos filhos de homens afetados são afetados.
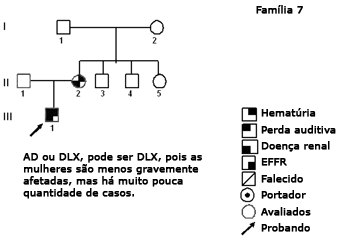
Figura 8. Heredograma família 7 - Provavelmente DLX. As mulheres são menos afetadas, mas há poucos casos.
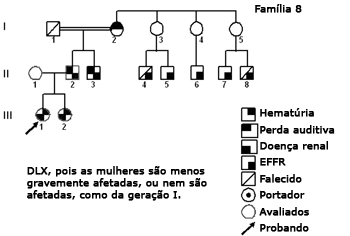
Figura 9. Heredograma família 8 - DLX, pois as mulheres são menos gravemente afetadas. Todas as filhas (III-1 e III-2) do afetado (II-2) são afetadas, característica da DLX.
2 - Análise estatística dos dados clínicos e da audição
Trinta e sete membros das oito famílias realizaram a avaliação aprovada pelo Comitê de Ética em Pesquisa da Instituição (Projeto nº 118/05). Foram excluídos os nove indivíduos normais.
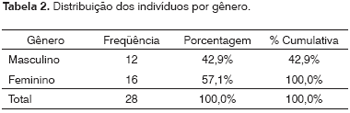
O gênero mais freqüente neste grupo foi o feminino (16/28; 57,1%) e o menos freqüente foi o masculino (12/28; 42,9%).
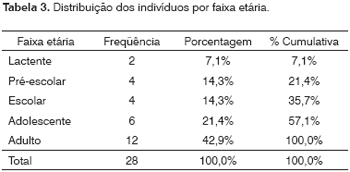
A faixa etária que mais predomina neste grupo de pacientes é a de adultos (12/28; 42,9%); a de pré-escolares, a de escolares e a de adolescentes perfazem 50,0% (14/28).
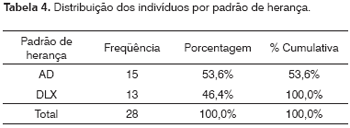
Os padrões de herança foram obtidos após a elaboração e análise dos heredogramas. Nesta amostra foram encontrados 46,4% de DLX e 53,6% de AD.
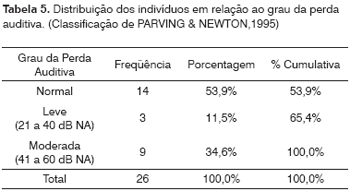
Doze pacientes tinham alteração da audição no exame audiométrico: 11,5% (3/26) com alteração leve e 34,6% (9/26) com alteração moderada. Foram excluídos da análise os lactentes (menores de dois anos), que não puderam realizar o exame de audiometria de tons puros. Em relação à faixa etária dos pacientes com alteração leve na audição (3/26): um é da faixa escolar (11 anos) e dois são adultos. Dos pacientes com alteração moderada (9/26) a distribuição etária é a seguinte: um escolar (oito anos), dois adolescentes (14 e 17 anos) e seis adultos. A PA ocorreu em 46,1% (12/26) dos indivíduos avaliados.Cinco pacientes tiveram indicação do uso de próteses auditivas, durante o estudo.
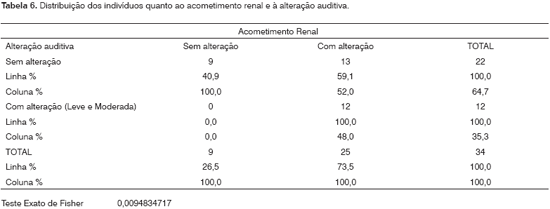
Analisamos os familiares sem comprometimento clínico e os comparamos aos que tinham alteração renal. Todos os que apresentavam alteração auditiva mostravam comprometimento renal. Existe associação entre acometimento renal e alteração auditiva (p= 0,009). RD%: 20,36 -61,45. 52% (13/25) dos pacientes em qualquer estágio da doença renal não exibiam alteração nos exames audiológicos. 48% (12/25) dos pacientes em qualquer estágio da doença renal manifestavam alteração leve ou moderada da audição. Desta análise foram excluídos os lactentes (2/37) que não fizeram a audiometria de tons puros e um caso (1/37) que não tinha exames laboratoriais de urina e não os realizou durante o estudo.
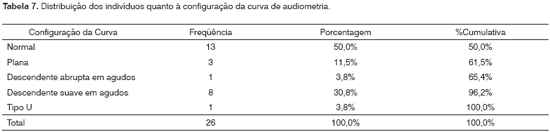
Apresentavam configuração normal da curva audiométrica 13 pacientes (50,0%) e 30,8% têm uma curva descendente suave em agudos (8/26). A configuração plana ocorreu em 11,5% (3/26). As configurações menos freqüentes foram: descendente abrupta em agudos e tipo U. Excluídos os dois lactentes.
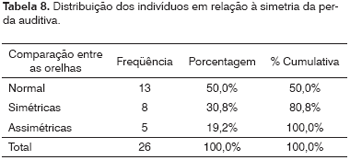
No grupo estudado, apesar de observarmos 30,8% (8/26) com perdas simétricas na comparação das duas orelhas, 19,2% (5/26) tinham perdas assimétricas. Foram excluídos os dois lactentes.
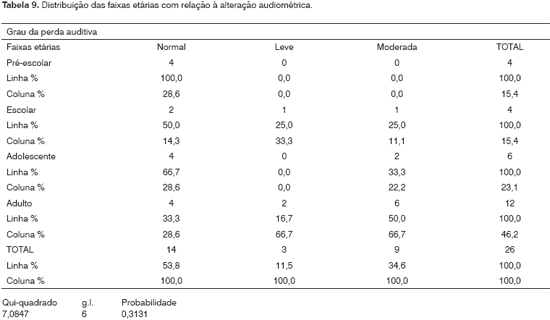
Observamos alteração auditiva nos escolares, nos adolescentes e nos adultos. Não foi detectada alteração nos pré-escolares. Foram excluídos os dois lactentes que não realizaram a audiometria de tons puros. Não houve correlação estatisticamente significante entre a alteração auditiva e a idade, nos pacientes estudados.
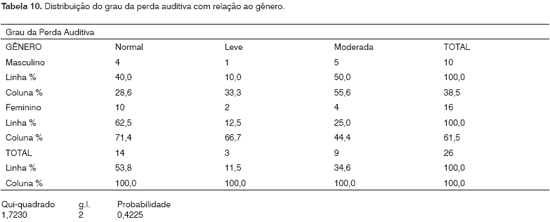
Neste grupo observamos 60,0% (6/10) dos homens com alteração na audição e 37,5% (6/16) das mulheres. Havia 10/16 (62,5%) mulheres sem alteração auditiva e 4/10 (40,0%) dos homens nas mesmas condições. Dois não realizaram a audiometria por não terem idade para o teste. A literatura relata que a perda auditiva ocorre, mais freqüentemente, em homens do que em mulheres.
DISCUSSÃO
Neste estudo tivemos uma freqüência maior de mulheres avaliadas (57,1%) em relação aos homens (42,9%). Na família dois constatamos quatro membros do sexo masculino falecidos e na família oito observamos três membros falecidos. Isto também poderia contribuir para uma menor participação dos homens no estudo (Tabela 2).
Os pré-escolares, escolares e adolescentes perfazem 50,0% (14/28) dos casos analisados (Tabela 3). Na população brasileira existe ainda um grande número de crianças, mas ocorreu um envelhecimento da população nos últimos anos, que modificou a distribuição das faixas etárias e a conformação da pirâmide. No censo demográfico do IBGE (2000) predominam os adultos e no grupo estudado também foi observado um predomínio de adultos (42,9%).
A elaboração dos heredogramas e a discussão destes com a equipe de genética contribuiu para a análise das famílias (Figuras 2 a 9). Observamos variabilidade intra e interfamiliar. A análise dos heredogramas permitiu supor a ocorrência de nova mutação na família sete (II-2).
A comprovação da alteração auditiva contribuiu para o diagnóstico de SA nas famílias que não haviam realizado biópsia renal, formando os critérios diagnósticos estritos. A biópsia renal pode ser duvidosa nas crianças e nas mulheres ou, ainda, arriscada nos casos de grave comprometimento renal.
Nesta amostra foram encontrados 46,4% de DLX e 53,6% de AD (Tabela 4). A expressiva participação dos indivíduos (9/28) da família um pode ter contribuído para este resultado. De acordo com a literatura,o padrão mais encontrado é o DLX e a análise do heredogramas sugere a forma autossômica dominante de SA.1 Em aproximadamente 85% dos heredogramas a doença é ligada ao X e as mutações identificadas no gene COL4A5.2 A classificação dos seis subtipos de SA9 é formada pelos padrões de herança AD e DLX. Os pacientes com SA autossômica dominante têm grande variabilidade clínica eo aprimoramento do diagnóstico molecular possa evidenciar que esta forma não é tão rara.3
Foram analisados os familiares sem comprometimento clínico e comparados aos que tinham alteração renal (Tabela 6). Todos os que apresentavam alteração auditiva mostravam comprometimento renal.Entre os que tinham avaliação audiométrica normal 59,1% apresentavam comprometimento renal. Existiu associação entre acometimento renal e alteração auditiva (p= 0,009) nos pacientes estudados. 48% (12/25) dos pacientes em qualquer estágio da doença renal tinham alteração leve ou moderada da audição. 52% (13/25) dos pacientes em qualquer estágio da doença renal não exibiam alteração nos exames audiológicos.
A alteração auditiva ocorre antes do estágio de falência renal, demonstrando que não é determinada pela uremia. Decorre das alterações das membranas basais nos órgãos potencialmente acometidos na SA, conforme os dados da literatura.10-13
A PA nunca é observada na ausência de sintomas renais.12
A progressão da alteração auditiva sugere pobre prognóstico renal, tornando evidente a estreita correlação, entre alteração auditiva e falência renal nos homens e mulheres.13,14
Entre os pacientes avaliados nesta amostra, doze pacientes tinham alteração da audição no exame audiométrico: 11,5% (3/26) com alteração leve e 34,6% (9/26) com alteração moderada (Tabela 5). Em relação à faixa etária dos pacientes com alteração leve na audição (3/26): um é da faixa escolar (11 anos) e dois são adultos. Dos pacientes com alteração moderada (9/26) a distribuição etária é a seguinte: um escolar (oito anos), dois adolescentes (14 e 17 anos) e seis adultos. Este comprometimento moderado pode decorrer da maior freqüência de adultos neste subgrupo. A PA ocorreu em 46,1% (12/26) dos indivíduos avaliados. É um achado extra-renal freqüente na SA.11 A perda auditiva tem sido relatada como um dos primeiros sintomas de SA6,14, podendo ocorrer já aos dez anos e ser socialmente insignificante até a segunda década de vida.12 A idade da perda de audição pode estar relacionada portanto ao tipo de mutação.2,15-17
Não houve correlação estatisticamente significativa entre a alteração auditiva e a idade nos pacientes estudados (Tabela 9). No entanto, na população geral não se observa alteração auditiva nos escolares, nos adolescentes e nos adultos, conforme o encontrado nesta análise. O teste do c2 (Qui-quadrado) é vulnerável e o tamanho da amostra somado às inúmeras possibilidades analisadas podem explicar o fato de não ter ocorrido associação entre a alteração auditiva e a idade.
Ainda neste grupo de estudo, 50,0% (13/26) dos pacientes apresentaram configuração normal da curva audiométrica e 30,8% (8/26) tiveram uma curva descendente suave em agudos (Tabela 7). A configuração plana ocorreu em 11,5% (3/26). As configurações menos freqüentes foram: descendente abrupta em agudos (3,8%) e tipo U (3,8%).
Três configurações audiométricas foram identificadas nas 51 orelhas que exibiam perda auditiva: forma escavada (47,1%), descendente (41,2%) e plana (11,7%).18
No grupo avaliado, observamos em 30,8% (8/26) perdas simétricas e em 19,2% perdas assimétricas (5/26) na comparação das duas orelhas (Tabela 8). A literatura relata que as perdas são simétricas.6,10,11 A amostra é pequena e não permite estabelecer uma comparação entre as orelhas.
Neste grupo observamos 60,0% (6/10) dos homens com alteração na audição e 37,5% (6/16) das mulheres. Houve 62,5% (10/16) das mulheres sem alteração auditiva e 40,0% (4/10) dos homens nas mesmas condições (Tabela 10). A literatura relata que a perda auditiva ocorre, mais freqüentemente, em homens do que em mulheres15, porém não observamos correlação entre alteração auditiva e gênero neste grupo de estudo.
O prognóstico mudou com o transplante renal, aumentando a longevidade dos pacientes com SA e falência renal. Tornando a investigação da evolução da PA e sua reabilitação, importantes para a qualidade de vida destes indivíduos. Há um vasto campo de pesquisa a ser desenvolvido, com animais geneticamente modificados ou com modelos animais, para a compreensão dos mecanismos fisiopatológicos envolvidos na alteração auditiva e na audição normal. Estes mesmos modelos podem ser empregados na pesquisa de novas terapias.
Não só os mecanismos fisiopatológicos da PASN na SA ainda merecem estudos adicionais, como também os sintomas clínicos, a alteração da audição e as relações genótipo-fenótipo. A atuação de grupos multidisciplinares, em diversos centros, trará informações adicionais sobre a doença, seu curso clínico, métodos diagnósticos e terapias. A PA na SA é ainda uma área excitante de investigação para geneticistas e otorrinolaringologistas.
CONCLUSÕES
- Os padrões de herança observados foram o AD e o DLX. A distribuição dos padrões de herança, neste estudo, não coincide com a descrita na literatura.
- A perda auditiva é um achado freqüente na SA.
- Todos os que apresentavam alteração auditiva mostravam comprometimento renal. Existe associação entre acometimento renal e alteração auditiva (p= 0,009).
- As configurações das curvas audiométricas mais encontradas, nesta amostra, foram: a descendente suave em agudos e a plana. As configurações menos freqüentes foram: descendente abrupta em agudos e tipo U.
- Não houve associação entre a alteração auditiva e a idade, neste estudo.
- Não observamos correlação entre alteração auditiva e gênero neste grupo de estudo.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Kashtan CE, Michael AF. Alport syndrome. Kidney Int 1996;50:1445-63.
2. Pirson Y. Making the diagnosis of Alport's syndrome. Kidney Int 1999;56:760-75.
3. Pescucci C, Mari F, Longo I, Vogiatzi P, Caselli R, Scala E, et al. Autosomal-dominant Alport syndrome: natural history of a disease due to COL4A3 or COL4A4 gene. Kidney Int 2004;65: 1598-603.
4. Barker DF, Hostikka SL, Zhou J, Chow LT, Oliphant AR, Gerken SC, et al. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 1990;248:1224-7.
5. Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer 2003;3:422-33.
6. Flinter FA, Cameron JS, Chantler C, Houston I, Bobrow M. Genetics of classic Alport's syndrome. Lancet 1988;1005-7.
7. Parving A, Newton V. Guidelines for description of inherited hearing loss. J Audiol Med 1995;4:2-5.
8. Liu X, Xu L. Nonsyndromic hearing loss: an analysis of audiograms. Ann Otol Rhinol Laryngol 1994;103:428-33.
9. Atkin CL, Hasstedt SJ, Menlove L, Cannon L, Kirschner N, Schwartz C, et al. Mapping of Alport syndrome to the long arm of the X chromosome. Am J Hum Genet 1988;42:249-55.
10. Atkin CL, Gregory MC, Border WA. Alport syndrome. In: Schrier RW, Gottschalk CW. Diseases of the kidney. 4th ed. Boston: Little, Brown 1988. p. 617-41.
11. Perkoff GT, Stephens FE, Dolowitz DA, Tyler FH. A clinical study of hereditary intersticial pyelonephritis. Arch Intern Med 1951;88:191-200.
12. Cassady G, Brown K, Cohen M, DeMaria W. Hereditary renal dysfunction and deafness. Pediatrics 1965;35:967-79.
13. Gluber M, Levy M, Broyer M, Naizot C, Gonzales G, Perrin D, et al. Alport's syndrome: a report of 58 cases and a review of the literature. Am J Med1981;70:493-505.
14. Di Paolo B, Vocino V, Bonomini M, Mouawad J, Albertazzi A. Significance of brain stem auditory evoked responses in Alport's syndrome. Contrib Nephrol 1990;80:88-94.
15. Wester DC, Atkin CL, Gregory MC. Alport syndrome: clinical update. J Am Acad Audiol 1995;6:73-9.
16. Barker DF, Pruchno CJ, Jiang X, Atkin CL, Stone EM, Denison JC, et al. A mutation causing Alport syndrome with tardive hearing loss is common in the Western United States. Am J Hum Genet 1996;58:1157-65.
17. Gross O, Netzer K-O, Lambrecht R, Seibold S, Weber M. Meta-analysis of genotype-phenotype correlation in X- linked Alport syndrome: impact on clinical counselling. Nephrol Dial Transplant 2002;17:1218-27.
18. Rintelmann FW. Auditory manifestations of Alport's disease syndrome. Trans Am Acad Ophthalmol Otolaryngol 1976;82:375-87.
1 Doutoranda em Medicina-Concentração em ORL-FCMSC-SP, Mestre em Medicina - Concentração em ORL-FCMSC-SP. Preceptora da Clínica de ORL - HSPM - SP.
2 Doutor em Medicina - Área de Concentração em Otorrinolaringologia, pela UNIFESP - EPM. Médico Chefe de Clínica Adjunto da Santa Casa de Misericórdia de São Paulo.
Trabalho realizado no Departamento de Otorrinolaringologia da Santa Casa de São Paulo.
Endereço para correspondência: Dra. Fátima Regina Abreu Alves - Av. Moema 801 São Paulo SP 04077-023. Tel: (0xx11) 5052-4344 - Fax: (0xx11)5052-0093 - E-mail: ffraa@terra.com.br
"Bolsista do CNPq - Brasil".
Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) da RBORL em 17 de junho de 2007. cod.4613
Artigo aceito em 22 de setembro de 2007.
Imprimir: ![]()