

Ano: 2008 Vol. 74 Ed. 5 - Setembro - Outubro - (15º)
Seção: Artigo Original
Páginas: 731 a 736
 PDF PT
PDF PT Rastreamento da mutação mitocondrial A1555G em pacientes com deficiência auditiva sensorioneural
Screening of the mitochondrial A1555G mutation in patients with sensorineural hearing loss
Autor(es): Luciano Pereira Maniglia1, Bruna Carolina Lemos Moreira2, Magali Aparecida Orate Menezes da Silva3, Vânia Belintani Piatto4, José Victor Maniglia5
Palavras-chave: aminoglicosídeos, deficiência auditiva, DNA mitocondrial, mutação.
Keywords: aminoglycosides, hearing loss, mitochondrial DNA, mutation.
Resumo:
A mutação mitocondrial A1555G é a principal alteração associada à surdez ocasionada pelo uso de aminoglicosídeos. Objetivo: Investigar a prevalência da mutação A1555G em pacientes com deficiência auditiva sensorioneural com e sem uso de antibióticos aminoglicosídeos. Material e Método: Estudo em amostras de 27 pacientes com surdez, como casos, e em 100 neonatos, com audição normal, como grupo controle. O DNA foi extraído de leucócitos de amostras de sangue e "primers" específicos foram utilizados para amplificar o gene do citocromo b e a região que abrange a mutação A1555G do DNA mitocondrial, usando as técnicas da Reação em Cadeia da Polimerase e do Polimorfismo no Comprimento de Fragmentos de Restrição. Desenho Científico: Estudo de casos em corte transversal. Resultados: A região do gene do citocromo b foi amplificada, sendo confirmada a presença do DNA mitocondrial em todas as 127 amostras do estudo. A mutação A1555G não foi identificada nos 27 pacientes com deficiência auditiva e no grupo controle (100 neonatos). Conclusões: Os resultados são concordantes com estudos que relatam que a mutação A1555G não é prevalente nas Américas. Há interesse na determinação da real prevalência dessa mutação e na investigação de outras mutações que possam ocasionar deficiência auditiva associada ou não ao uso de aminoglicosídeos na população brasileira.
Abstract:
The A1555G mitochondrial mutation is the main alteration associated with aminoglycoside-induced deafness. Aim: to investigate the prevalence of the A1555G mutation in patients sensorineural hearing loss patients with and without aminoglycosides antibiotic use. Material and Method: a study of 27 cases with deafness as the sample, and 100 neonates with normal hearing as the control group. DNA was extracted from blood leukocyte samples, and specific oligonucleotide primers were designed to amplify the cytochrome b gene and the region which encloses the A1555G mutation of the mitocondrial DNA using the polymerase chain reaction and restriction fragment length polymorphism. Design: a cross-sectional case study. Results: a region of the cytochrome b gene was amplified and the presence of the mtDNA was confirmed in all of the 127 cases. The A1555G mutation was not found in any of the 27 patients with hearing loss or the control group with 100 neonates. Conclusion: the results agree with studies stating that the A1555G mutation is not prevalent in the Americas. There is interest in establishing the real prevalence of this mutation and to investigate other mutations that may cause hearing loss, associated or not with the use of aminoglycosides, in the Brazilian population.
![]()
INTRODUÇÃO
Nos países desenvolvidos, estima-se que a cada 750 nascimentos, uma criança apresenta deficiência auditiva do tipo sensorioneural e, também uma, em cada 1000 crianças, torna-se surda antes de alcançar a idade adulta. A prevalência continua a aumentar e alcança índices de 50% em octogenários1. Aproximadamente, 60% de todas as causas de deficiência auditiva pré-lingual podem ser atribuídas a fatores genéticos. Desse modo, a etiologia genética passa a ter cada vez mais relevância nos casos de deficiência auditiva e/ou surdez. Os 40% restantes estão entre as mais diversas etiologias2. Existem três formas identificáveis de padrão de herança para a deficiência auditiva hereditária: autossômica recessiva, autossômica dominante e ligada ao cromossomo X. Cerca de 75% a 85% dos casos de deficiência auditiva pré-lingual não-sindrômica manifestam-se como formas autossômicas recessivas. Formas autossômicas dominantes correspondem a cerca de 15% a 25% dos casos e os restantes 1% a 3%, são heranças mendelianas ligadas ao cromossomo X. Também são descritas formas herdadas exclusivamente da mãe, correspondendo à herança mitocondrial, em cerca de 0,5% a 1% das causas genéticas de deficiência auditiva sensorioneural, mas podendo ocorrer mutações espontâneas2,3. A relação entre doença mitocondrial e deficiência auditiva foi estabelecida em 1986, pelo estudo de um paciente com miopatia mitocondrial e deficiência auditiva4.
Mutações mitocondriais, especialmente nos genes 12S rRNA e tRNASer(UCN), são causas importantes de deficiência auditiva sensorioneural não-sindrômica, em algumas populações5. Dentre todas essas mutações, uma substituição de bases nitrogenadas A->G, na posição 1555 no gene 12S rRNA do DNA mitocondrial, é de particular interesse como a principal causa de deficiência auditiva relacionada ao uso de antibióticos aminoglicosídeos. Foi identificada, em 1993, em um estudo que a descreveu como uma mutação no rRNA mitocondrial que ocasionava deficiência auditiva não-sindrômica, sendo a primeira análise molecular genética relacionada à ototoxicidade induzida por aminoglicosídeos6.
Vários estudos têm proposto que a mutação A1555G, pela substituição das bases nitrogenadas, cria um novo par de bases C-G tornando, a estrutura secundária do gene 12S rRNA humano, estritamente semelhante à região correspondente do gene 16S rRNA da E. coli sendo, essa, referida como a região decodificante do rRNA bacteriano e um importante local de ação dos antibióticos aminoglicosídeos6,7. Sendo assim, a ligação de aminoglicosídeos à região decodificante resulta em erros na tradução protéica com concomitante morte bacteriana e, quando há a mutação, geralmente em homoplasmia, pelas alterações no gene 12S rRNA humano, que o tornam de estrutura semelhante ao rRNA bacteriano, ocorre também a ligação dos aminoglicosídeos no DNA mitocondrial das células cocleares, potencializando seu efeito tóxico na orelha interna, ocasionando deficiência auditiva. Portanto, esses estudos demonstram o modo de reconhecimento, altamente específico, entre moléculas de RNA e aminoglicosídeos7.
Em contraste as mutações mitocondriais sindrômicas que, usualmente, afetam somente uma fração de todas as moléculas do DNA mitocondrial (heteroplasmia), as mutações associadas à deficiência auditiva sensorioneural não-sindrômica são, freqüentemente, homoplásmicas e o fenótipo difere consideravelmente entre membros de uma mesma família, variando de grau profundo à audição completamente normal. Vários fatores, tais como genes nucleares, haplótipos do DNA mitocondrial, fatores ambientais ou efeitos tecido-específicos atuam independentemente ou em combinação, podendo influenciar a expressão clínica, mas estudos não têm identificado genes modificadores nucleares ou correlação entre a mutação e haplótipos mitocondriais. O único fator ambiental conhecido em afetar a mutação A1555G é o grupo de antibióticos aminoglicosídeos8.
Embora as mutações mitocondriais tenham grande importância na etiologia da deficiência auditiva sensorioneural, existem poucos estudos em países em desenvolvimento, especialmente no Brasil. Sendo assim, o presente estudo teve como objetivo investigar a prevalência da mutação mitocondrial A1555G, em amostra de pacientes com deficiência auditiva sensorioneural não sindrômica com e sem exposição aos aminoglicosídeos.
CASUÍSTICA E MÉTODO
No período de Setembro a Outubro de 2006, foi realizado um estudo de corte transversal no qual foram estudados 20 casos-índice com deficiência auditiva sensorioneural não-sindrômica (12 do sexo masculino e 8 do sexo feminino), com idade entre 1 e 37 anos, os quais já haviam sido anteriormente submetidos à análise molecular do gene GJB2 e da mutação D(GJB6-D13S1830) e não apresentaram qualquer alteração molecular9. Os casos-índice estudados eram constituídos por 16 casos esporádicos (único caso na família) e 4 familiais (casos familiais). Destes casos familiais, foram avaliados também os parentes com deficiência auditiva (2 do sexo masculino e 5 do sexo feminino), com idade entre 5 e 45 anos. Portanto, foi estudado um total de 27 pacientes com deficiência auditiva.
Cada paciente foi submetido à completa anamnese para investigar idade de início da deficiência auditiva, presença ou não de outros casos na família, uso ou não de drogas ototóxicas (aminoglicosídeos) e excluir a possibilidade de outras causas ambientais: infecções materno-fetais, complicações perinatais, meningites, trauma acústico, casamento consangüíneo. Os exames físicos, otorrinolaringológico e sistêmico e exames complementares foram realizados para se excluir sinais sugestivos de formas sindrômicas de deficiência auditiva (especialmente dismorfismo crânio-facial, alterações tegumentares, anomalias de origem branquial, cardíaca, tireoidianas, distúrbios da visão, etc.). Além disso, os pacientes foram submetidos à avaliação oftalmológica (incluindo fundoscopia), testes vestibulares e tomografia computadorizada de osso temporal. Portanto, toda a avaliação clínica foi realizada para se excluir pacientes com deficiência auditiva causada por fatores ambientais, exceto o uso de aminoglicosídeos, por consangüinidade, malformações congênitas de orelha interna ou por síndromes genéticas. Os pacientes foram audiologicamente testados por audiometria de tom puro e incluídos aqueles com deficiência auditiva sensorioneural não-sindrômica classificada como leve (25-40 dB), moderada (41-60 dB), grave (61-80 dB) ou profunda (>81 dB)10.
Os pacientes incluídos foram identificados como Grupo Deficiente Auditivo (GDA) sendo dividido em dois subgrupos:
. Grupo GDA I - pacientes que fizeram uso de antibióticos aminoglicosídeos (n=13).
. Grupo GDA II - pacientes que não fizeram uso de antibióticos aminoglicosídeos (n=14).
Como Grupo Controle (GC) foram incluídos no estudo 100 neonatos, com teste normal de emissões otoacústicas realizado até 3 dias após o nascimento a termo e com índice de Apgar >7 no 1° minuto.
Foram coletados 4,0mL de sangue total (de veia periférica no Grupo GDA e de cordão umbilical, após sua ligadura, no Grupo Contrrole) em um tubo Vacutainer? contendo anticoagulante (EDTA), com prévia obtenção do termo de consentimento livre e esclarecido dos pacientes ou responsáveis. O protocolo para este estudo foi aprovado pelo Comitê de Ética em Pesquisa (CEP) da Instituição (Protocolo nº 2784/2006). O DNA genômico foi extraído das amostras de sangue de ambos os Grupos usando o GFXTM Genomic Blood DNA Purification Kit (Amersham Pharmacia Biotech Inc.), de acordo com o protocolo do fabricante. Para se detectar a mutação A1555G11, fragmentos do DNA mitocondrial, que abrangem a região da mutação, foram amplificados pela técnica da Reação em Cadeia da Polimerase (PCR - Polimerase Chain Reaction), no termociclador Applied Biosystems - GeneAmp PCR System 9700Ò. Para esta reação foi sintetizado um par de iniciadores ou "primers". A partir destes, os desoxinucleotídeos trifosfato (dNTPs) são incorporados, iniciando a amplificação do DNA, respeitando-se a complementaridade de bases (A-T/C-G) sendo obtida, por fim, a amplificação do DNA mitocondrial. As seqüências dos oligonucleotídeos dos "primers" e as condições para a reação da PCR foram de acordo com as descritas na literatura11.
Como produto da reação da PCR, foi amplificado um fragmento de 643 pb o qual, posteriormente, foi submetido à análise de restrição pela técnica do Polimorfismo no Comprimento de Fragmentos de Restrição (RFLP - Restriction Fragment Lengt Polymorphism), utilizando-se a enzima BsmAI11 (New England Biolabs)Ò, por 2 horas a 55°C. A digestão enzimática do fragmento de amostras normais para a mutação A1555G produz dois fragmentos de 413 pb e 230 pb, devido o reconhecimento do sítio de restrição da enzima BsmAI. As amostras com a mutação produzem apenas o fragmento de 643 pb, pois não há o reconhecimento do sítio da enzima, devido a substituição das bases nitrogenadas A->G, na posição 1555 do DNA mitocondrial.
Foi também sintetizado um outro par de "primers" para amplificação de determinada região do citocromo b12. Essa região específica, uma área altamente conservada do genoma mitocondrial, serve como controle interno de amplificação para verificar a presença do DNA mitocondrial nas amostras. O tamanho esperado do fragmento amplificado pela PCR usando-se os "primers" CitbF e CitbR foi de 161 pb. As seqüências dos oligonucleotídeos dos "primers" e as condições para a reação da PCR foram de acordo com as descritas na literatura12.
Os dois pares de "primers" utilizados para as PCR abrangem regiões do "Human Mitochondrial DNA Revised Cambridge Reference Sequence"13.
Os produtos de ambas as reações, PCR e RFLP, foram analisados por eletroforese em gel de agarose 2% em tampão TBE 1X, contendo brometo de etídio, na concentração de 0,5mg/mL, submetidos à iluminação ultravioleta, para confirmar o sucesso da reação e o gel, fotodocumentado.
Análise estatística:
Foram avaliados 100 recém-nascidos, em estudo piloto, para estimativa da proporção (p) de prováveis portadores da mutação A1555G nesta amostra inicial. A esta proporção (p), após ter sido determinada, foi aplicada a fórmula estatística "dimensionamento de amostra com população conhecida", obtendo-se o tamanho da amostra final (n) necessária para representar estatisticamente a população total (Np) de recém-nascidos, nascidos no período determinado para a realização da pesquisa (Np=352 neonatos). Para o cálculo da amostra final (n), foi utilizada a referida fórmula estatística, com os seguintes parâmetros: p=0,00 (estimado pela amostra piloto); q=1,00; zc=3,00 (99,74% de confiabilidade); e=0,03 (3% de erro de estimativa); Np=352 (tamanho da população no período do estudo).
Fórmula: n = zc2 × p × q × Np
e2 × (Np - 1) zc2 × p × q
Os resultados do estudo foram expressos em porcentagens.
RESULTADOS
A amplificação de região do gene do citocromo b foi obtida após reação da PCR visualizando-se o fragmento correspondente de 161 pb confirmando, portanto, a presença do DNA mitocondrial em todas as 127 amostras do estudo - 100% (Grupos GDA I e II - n=27 e Grupo Controle - n=100).
A reação da PCR permitiu a amplificação do fragmento de 643 pb do DNA mitocondrial, que abrange a região da mutação, em todas as amostras analisadas (100%) dos Grupos GDA I e II (n=27) e Controle (n=100).
A mutação mitocondrial A1555G não foi identificada nas amostras do estudo (100%) pois, após a digestão enzimática, observou-se os fragmentos de 413 pb e 230 pb, os quais se amplificam na ausência da mutação, em todas as amostras de ambos os grupos (Grupos GDA I e II - n=27 e Grupo Controle - n=100).
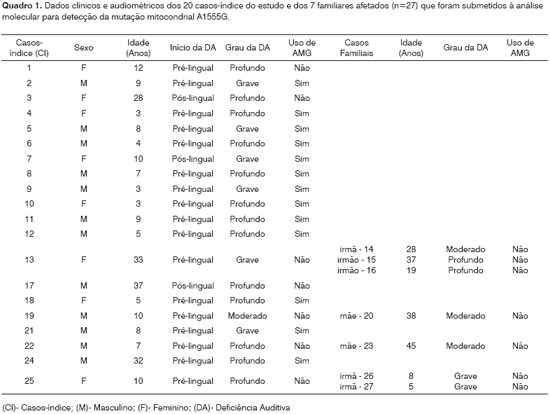
Os dados clínicos e audiométricos obtidos dos casos-índice tais como sexo, idade, época de início e grau de perda da deficiência auditiva, uso ou não de aminoglicosídeos e casos familiais, são descritos no Quadro 1.
DISCUSSÃO
Em continuidade às pesquisas que identificaram que mutações no gene da conexina 26 (Cx26) ocasionam deficiência auditiva em uma significante proporção de casos, em populações de diversos países europeus, a descoberta de outras mutações tornou possível a identificação etiológica molecular em muitos pacientes propiciando, além da reabilitação precoce, o aconselhamento genético. Entretanto, tem sido interessante observar que a freqüência de genes e mutações que ocasionam deficiência auditiva tem variado, significantemente, entre as populações. A mutação 35delG, no gene da Cx26, por exemplo, tem uma alta prevalência entre surdos de origem européia9,14 enquanto é praticamente ausente em japoneses, coreanos ou mongolianos15,16.
Do mesmo modo, a freqüência de mutações mitocondriais também difere entre as diversas populações, sendo a mutação A1555G encontrada, principalmente, em pacientes com deficiência auditiva de transmissão materna, embora tem sido identificada, esporadicamente, em pacientes com herança autossômica recessiva. Em ambos os modos de transmissão evidencia-se grandes diferenças nas origens étnicas, conforme estudos recentes que a apontam como uma causa freqüente de deficiência auditiva hereditária não-sindrômica, associada ou não ao uso de antibióticos aminoglicosídeos em populações do continente asiático11,17-21 de países árabes22 e da África do Sul23, sendo rara na maior parte da população européia e americana24,25.
Estudo realizado em 32 asiáticos com deficiência auditiva e em 100 controles Americanos (63 surdos e 37 ouvintes) encontrou a mutação em 87,5% do grupo asiático surdo, não a encontrando em nenhum dos indivíduos do grupo controle, concluindo que a mutação não é prevalente em população de origem não-asiática25. Esse resultado foi confirmado em estudo na Grécia, no qual a mutação não foi encontrada em 106 casos26 e em 202 pacientes com deficiência auditiva não-sindrômica de início precoce no Reino Unido5. Da mesma forma, no presente estudo, não foi encontrada a mutação mitocondrial A1555G nos Grupos GDA (Grupo I - 13 casos-índice que fizeram uso de aminoglicosídeos e Grupo II - 14 casos-índice que não fizeram uso de aminoglicosídeos) e Controle (100 neonatos com teste de emissões otoacústicas normal). Este resultado demonstra que a mutação não é uma causa comum de deficiência auditiva na presente amostra estudada.
Nos EUA foram realizados dois rastreamentos da mutação em neonatos. No primeiro, utilizando sangue de cordão umbilical de 1773 recém-nascidos, foi observado apenas um portador da mutação27. No segundo rastreamento em 25 neonatos, com alterações no teste de emissões otoacústicas, não foi encontrada a mutação28. Na Argentina, rastreamento realizado em 300 recém-nascidos e em 712 indivíduos ouvintes, também não foi encontrada a mutação A1555G29. Esses estudos referem a baixa prevalência nas populações americanas, mas não descartam a necessidade da investigação molecular em testes de rastreamento neonatais, principalmente, em países nos quais há uso rotineiro de aminoglicosídeos.
A metodologia do presente estudo foi semelhante à descrita na literatura11, incluindo a amplificação de uma específica região do gene do citocromo b, uma área altamente conservada do genoma mitocondrial, que serviu com um controle de amplificação para se verificar a presença do DNA mitocondrial em todas as amostras analisadas12. Apesar do número de casos, tanto do Grupo GDA como do Grupo Controle diferirem de alguns estudos da literatura e, mesmo não tendo sido encontrada a mutação em ambos os grupos, confirmou-se os resultados de estudos que também não a encontraram em grupos étnicos não-asiáticos ou árabes24-29.
No Brasil foi realizado um único estudo em 5 famílias com casos de deficiência auditiva, no qual foi determinada a prevalência de 2% (4 casos) da mutação. Desses casos positivos, apenas 1 estava associado a uma suposta exposição aos aminoglicosídeos e não foi encontrada a mutação no grupo controle constituído por afro-brasileiros, "brancos" e de origem asiática (japoneses ou chineses)30. Tais resultados, diferentes do presente estudo, podem ser explicados pela composição étnica altamente heterogênea da população brasileira, devido à miscigenação entre vários grupos étnicos, podendo resultar, portanto, em diferentes índices de prevalência nas várias regiões brasileiras.
Muitas mutações podem contribuir para a deficiência auditiva e a mutação mitocondrial A1555G, relacionada ao uso de antibióticos aminoglicosídeos, pode ser apenas uma delas17,30. A identificação de causas subjacentes da deficiência auditiva ocasionada por exposição aos aminoglicosídeos é fundamental para auxiliar na terapêutica, principalmente em UTI neonatal, além de propiciar o aconselhamento genético e reabilitação precoce, permitindo a inclusão ou re-inclusão destes pacientes em suas atividades sociais e profissionais.
CONCLUSÃO
As técnicas moleculares PCR/RFLP, com o protocolo utilizado no estudo, são métodos de fácil execução para o rastreamento da mutação A1555G colaborando para a investigação molecular da deficiência auditiva.
Há considerável interesse na determinação da real prevalência, na população brasileira, da mutação mitocondrial A1555G e na investigação de outras mutações que possam ocasionar deficiência auditiva associada ou não ao uso de aminoglicosídeos, por meio dos testes moleculares, conforme realizado no presente estudo, os quais podem ser um valioso complemento aos rastreamentos audiométricos neonatais.
AGRADECIMENTOS
Agradecemos aos pacientes e aos pais dos recém-nascidos sem cujo consentimento e cooperação este estudo não seria possível. Esta contribuição é extremamente importante para permitir a continuidade das pesquisas científicas a fim de proporcionar um futuro melhor às crianças brasileiras.
À amiga e tradutora, Cecília Meneguette Ferreira, por seu inestimável auxílio.
REFERÊNCIAS BIBLIOGRÁFICAS
1. American College of Medical Genetics. Genetics Evaluation Guidelines for the Etiologic Diagnosis of Congenital Hearing Loss. Genet Med 2002;4:162-71.
2. Mustafa T, Arnos KS, Pandya A. Advances in hereditary deafness. Lancet 2001;358:1082-90.
3. Denoyelle F, Marlin S, Petit C, Garabédian E-N. Surdités neurosensorielles d´origine génétique. Rev Prat 2000;50:146-9.
4. Petty RKH, Harding AE, Morgan-Hughes JA. The clinical features of mitochondrial myopathy. Brain 1986;109:915-38.
5. Hutchin TP, Thompson KR, Newton V, Bitner-Glindzicz M, Mueller RF. Prevalence of mitochondrial DNA mutations in childhood/congenital onset non-syndromal sensorineural hearing impairment. J Med Genet 2001;38:229-31.
6. Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu WQ, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet 1993;4:289-94.
7. Hamasaki K & Rando RR. Specific binding of aminoglycosides to a human rRNA construct based on a DNA polymorphism wich causes aminoglycoside-induced deafness. Biochemistry 1997;36:12323-8.
8. Giordano C, Pallotti F, Walker WF, Checcarelli N, Musumeci O, Santorelli F, et al. Pathogenesis of the deafness-associated A1555G mitochondrial DNA mutation. Biochem Biophys Res Commun 2002;293:521-9.
9. Belintani VP, Goloni-Bertollo EM, Sartorato EL, Maniglia JV. Prevalence of the GJB2 mutations and the del(GJB6-D13S1830) mutation in Brazilian patients with deafness. Hear Res 2004;196:87-93.
10. World Health Organization. Report of the informal working group on prevention of deafness and hearing impairment programme planning. Geneva: WHO, 1991. 22p
11. Noguchi Y, Yashima T, Ito T, Sumi T, Tsuzuku T, Kitamura K. Audiovestibular findings in patients with mitochondrial A1555G mutation. Laryngoscope 2004;114:344-8.
12. Bai U, Seidman MD, Hinojosa R, Quirk WS. Mitochondrial DNA deletions associated with aging and possibly presbicusis: a human archival temporal bone study. Am J Otol 1997;18:449-53.
13. Andrews RM, Kubacha I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N, 1999. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999;23(2), 147.
14. Tekin M, Akar N, Cin S, Blanton SH, Xia XJ, Liu XZ, et al. Connexin 26 (GJB2) mutations in the Turkish population: implications for the origin and high frequency of the 35 delG mutation in Caucasians. Human Genet 2001;108:385-9
15. Abe S, Usami S, Shinkawa H, Kelley PM, Kimberling WJ. Prevalent connexin 26 gene (GJB2) mutations in Japanese. J Med Genet 2000;37:41-3.
16. Pandya A, Tekin M, Erdenetungalag R, Xia A, Dangaasuren B, Radnaabazar J, et al. A unique spectrum of alterations in the Cx26 gene in deaf probands from Mongolia. Eur J Hum Genet 2001;9:388.
17. Matsunaga T, Kumanomido H, Shiroma M, Ohtsuka A, Asamura K, Usami S. Deafness due to A1555G mitochondrial mutation without use of aminoglycoside. Laryngoscope 2004;114:1085-90.
18. Matsunaga T Kumanomido H, Shiroma M, Goto Y, Usami S. Audiological features and mitochondrial DNA sequence in a large family carrying mitochondrial A1555G mutation without use of aminoglycoside. Ann Otol Rhinol Laryngol 2005;114:153-60.
19. Tamagawa Y, Kitamura K, Ishida T, Hagiwara H, Abe K, Nishizawa M. Mitochondrial DNA mutation at nucleotide 1555 in a patient with bilateral sensorineural hearing loss of unknown etiology. Acta Otolaryngol 1996;116:796-8.
20. Malik SG, Pieter N, Sudoyo H, Kadir A, Marzuki S. Prevalence of the mitochondrial DNA A1555G mutation in sensorineural deafness in Island Southeast Asia. J Hum Genet 2003;48:480-3.
21. Pandya A, Xia X, Radnaabazar J, Batsuuri J, Dangaansuren B, Fischel-Ghodsian N, Nance WE. Mutation in the mitochondrial 12S rRNA gene in two families from Mongolia with matrilineal aminoglycoside ototoxicity. J Med Genet 1997;34:169-72.
22. Tekin M, Duman T, Bogoçlu G, Incesulu A, Comak E, Fitoz S, et al. Frequency of mtDNA A1555G and A7445G mutations among children with prelingual deafness in Turkey. Eur J Pediatr 2003;162:154-8.
23. Gardner JC, Goliath R, Viljoen D, et al. Familial streptomycin ototoxicity in a South African family: a mitochondrial disorder. J Med Genet 1997;34:904-6.
24. Hutchin T, Cortopassi G. Mitochondrial DNA haplotype predicts deafness risk. AM J Med Genet 1995;60:592
25. Usami S, Abe S, Kasai M, Shinkawa H, Moeller B, Kenyon JB, et al.. Genetic and clinical features of sensorineural hearing loss associated with the 1555 mitochondrial mutation. Laryngoscope 1997;107:438-90.
26. Kleomitis E, Iliadis T, Voyiatzis N, Economides J, Neou P, Apostolopoulos N, et al. Analysis of the A1555G mtDNA mutation in Greek patients with sensorineural deafness. Am J Hum Genet 2000;67:1879.
27. Tang HY, Hutcheson E, Neil S Drummond-Borg M, Sper M, Alford RL. Genetic susceptibility to aminoglycoside ototoxicity: How many are at risk? Genet Med 2002;4:336-45.
28. Dent KM, Kenneson A, Palumbos JC, Maxwell S, Eichwald J, White K, et al. Methodology of a multistate study of congenital hearing loss: preliminary data from Utah newborn screening. Am J Med Genet C Semin Med Genet 2004;125:28-34.
29. Gravina LP, Foncuberta ME, Estrada RC, Barreiro C, Chertkoff L. Carrier frequency of the 35delG and A1555G deafness mutations in the Argentinean population impact on the newborn hearing screening. Int J Pediatr Otorhinolaryngol 2007;4:639-43.
30. Abreu-Silva RS, LezIrovitz K, Braga MCC, Spinelli M, Pirana S, Della-Rosa VA, et al. Prevalence of the A1555G (12S rRNA) and tRNASer (UCN) mitochondrial mutations in hearing-impaired Brazilians patients. Braz J Med Biol Res 2006;39:219-26.
1 Pós-graduando mestrado, Docente do Departamento de Otorrinolaringologia e Cirurgia de Cabeça e Pescoço da Fac. Medicina S. J. Rio Preto SP FAMERP.
2 Graduanda em Medicina da Faculdade de Medicina de São José do Rio Preto SP FAMERP.
3 Mestre, Chefe do Serviço de Fonoaudiologia do Departamento de Otorrinolaringologia e Cirurgia de Cabeça e Pescoço da Fac. Medicina S. J. Rio Preto SP FAMERP.
4 Doutorado, Professor Adjunto.
5 Livre-docente, Chefe do Departamento de Otorrinolaringologia e Cirurgia de Cabeça e Pescoço da Fac. Medicina S. J. Rio Preto SP FAMERP. Faculdade de Medicina de São José do Rio Preto SP FAMERP.
Endereço para correspondência: Vânia Belintani Piatto - Rua Santina Figliagi Ceccato 450 ap. 23-A São José do Rio Preto SP 15035-180
Tel. (0xx17) 3201-5747 - E-mail: vbpiatto@gmail.com ou vabp@ig.com.br
BIC - FAMERP (Bolsa de Iniciação Científica - FAMERP).
Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) da RBORL em 2 de maio de 2007. cod. 4488
Artigo aceito em 28 de junho de 2007.
Imprimir: ![]()