

Ano: 2007 Vol. 73 Ed. 6 - Novembro - Dezembro - (9º)
Seção: Artigo Original
Páginas: 777 a 783
 PDF PT
PDF PT Correlação entre dados audiométricos e mutação 35delG em dez pacientes
Correlation between audiometric data and the 35delG mutation in ten patients
Autor(es): Vânia Belintani Piatto 1, Otávio Augusto Vasques Moreira 2, Magali Aparecida Orate Menezes da Silva 3, José Victor Maniglia 4, Márcio Coimbra Pereira 5, Edi Lúcia Sartorato 6
Palavras-chave: análise molecular, audiometria, deficiência auditiva, mutação 35delg
Keywords: molecular analysis, audiometry, hearing loss, 35delg mutation
Resumo:
Mutações no gene da conexina 26 parecem ser extremamente comuns na gênese da surdez hereditária não-sindrômica, especialmente, a mutação 35delG, mas ainda há poucos estudos que descrevem as características audiométricas dos pacientes portadores dessas mutações. Objetivo: Analisar as características audiométricas em pacientes com mutações no gene da conexina 26 para se delinear uma correlação genótipo-fenótipo. Casuística e Método: Foram avaliadas audiometrias tonal de 33 casos-índice com surdez sensorioneural não-sindrômica e de 8 familiares afetados. Testes moleculares específicos foram realizados para analisar mutações no gene da conexina 26. Forma de Estudo: Estudo de casos, retrospectivo, em corte transversal. Resultados: Foram encontradas as prevalências de 27,3% da mutação 35delG nos casos-índice e de 12,5% nos familiares afetados. Em relação aos graus de perda, foram encontrados, 41,5% dos pacientes com grau profundo, 39,0% com grau grave e 19,5% com grau moderado com, os pacientes homozigotos e heterozigotos para 35delG, predominando nos graus moderado-grave. Conclusão: Estes resultados sugerem que os dados audiométricos, associados ao diagnóstico molecular para a surdez, permitiram delinear uma correlação genótipo-fenótipo em dez pacientes com a mutação 35delG. Mas é necessário estudo multicêntrico para se verificar a real expressão fenotípica na população brasileira relacionada à mutação 35delG.
Abstract:
Mutations in the connexin 26 gene seem to be extremely common in non-syndromic hereditary deafness genesis, especially the 35delG, but there are still only a few studies that describe the audiometric characteristics of patients with these mutations. Aim: to analyze the audiometric characteristics of patients with mutations in the connexin 26 gene in order to outline genotype-phenotype correlation. Materials and Methods: Tonal audiometries of 33 index cases of non-syndromic sensorineural hearing loss were evaluated and eight affected relatives. Specific molecular tests were carried out to analyze mutations in the connexin 26 gene. Experiment Design: Retrospective, cross-sectional study. Results: A 27.3% prevalence of mutation 35delG was found in the index cases and 12.5% among the relatives affected. In relation to hearing loss degree, 41.5% of the patients were found with profound hearing loss, 39% with severe HL and 19.5% with moderate HL with homozygote and heterozygote patients for the 35delG predominating in the severe-moderate hearing losses. Conclusion: Our results suggest that the audiometric data associated with the molecular diagnose of hearing loss helped us to outline a genotype-phenotype correlation in ten patients with 35delG mutation. However, it is still necessary to run multicentric studies to verify the real phenotypic expression in the Brazilian population, as far as the 35delG mutation is concerned.
![]()
INTRODUÇÃO
Nos países desenvolvidos sabe-se que, de cada 750 nascimentos, uma criança tem a possibilidade de apresentar deficiência auditiva do tipo sensorioneural e estima-se que, uma em cada 1000 crianças seja afetada por surdez grave ao nascer ou até o término do período pré-lingual1. Aproximadamente 60% de todas as causas de surdez pré-lingual podem ser atribuídas a fatores genéticos. Desse modo, a etiologia genética passa a ter cada vez mais relevância nos casos de deficiência auditiva e/ou surdez. Os 40% restantes estão entre as mais diversas etiologias2. Existem três formas identificáveis de padrão de herança para a surdez hereditária: autossômica recessiva, autossômica dominante e ligada ao cromossomo X. De 75% a 85% dos casos de surdez pré-lingual não-sindrômica manifestam-se como formas autossômicas recessivas. Formas autossômicas dominantes correspondem a cerca de 15% a 25% dos casos e os restantes 1% a 3%, são heranças mendelianas ligadas ao cromossomo X. Também são descritas formas herdadas exclusivamente da mãe, correspondendo à herança mitocondrial, associadas ou não à herança autossômica dominante2,3.
As formas autossômicas recessivas, em termos fenotípicos, são mais graves, sendo responsáveis por aproximadamente todas as formas de surdez congênita. Essas, por sua vez, são ocasionadas, em grande parte, por defeitos cocleares, levando à deficiência auditiva sensorioneural. As formas autossômicas dominantes parecem ter maior contribuição nos casos de surdez pós-lingual sendo, geralmente, progressivas e o déficit, na maioria dos casos, do tipo condutivo ou misto (condutivo e sensorioneural)4,5.
Mutações no gene da conexina 26 ou GJB2 - Gap Junction Protein Beta 2, localizado no braço longo do cromossomo 13 (13q11-12), parecem ser extremamente comuns na gênese da surdez hereditária não-sindrômica, sendo responsáveis por 34% a 50% das formas de surdez sensorioneural autossômica recessiva (DFNB1) e por 10% a 37% dos casos esporádicos6-10.
A deleção de uma guanina, de uma seqüência de 6 guaninas que se estende da posição 30 à posição 35 no gene da conexina 26 (GJB2), é a mutação que ocorre em 60% a 80% dos casos. Essa deleção do nucleotídeo pode ocorrer na posição 35 (35delG) ou na posição 30 (30delG) do gene, mas a deleção na posição 35 (35delG), geneticamente relacionada ao cromossomo 13, é a mais freqüente, variando aproximadamente em 50% a 85% dos casos, em pacientes da Itália, Espanha, Israel com surdez não-sindrômica7,11-13. Mutações no gene GJB2 foram encontradas em 22% das famílias testadas com pelo menos um paciente com deficiência auditiva e em 11,5% dos casos, nos quais a etiologia ambiental não foi completamente excluída, em estudo em pacientes brasileiros14. Nos casos onde ocorre essa mutação no gene da conexina 26, o produto do gene, a proteína também denominada conexina, passa a não exercer suas funções corretamente, estando a cóclea estruturalmente normal15.
Embora o gene GJB2 tenha grande importância na etiologia da deficiência sensorioneural não-sindrômica, ainda há poucos estudos que descrevem as características audiométricas dos pacientes com mutações no gene GJB2 associadas à deficiência auditiva, especialmente, no Brasil. De acordo com esses estudos, a deficiência auditiva, nos pacientes homozigotos para mutações no gene GJB2 e principalmente nos homozigotos para 35delG, se caracteriza por ser pré-lingual, atingir todas as freqüências, não-progressiva, variar de grau moderado a profundo, mesmo entre irmãos deficientes de uma mesma família e não estar associada a alterações vestibulares e anormalidades radiológicas da orelha interna16,17. O presente estudo tem como objetivo analisar as características audiométricas em pacientes com mutações no gene da conexina 26 para se delinear uma correlação genótipo-fenótipo.
CASUÍSTICA E MÉTODO
No período de Março a Junho de 2000, foi realizado um estudo de corte transversal, no qual foram estudados 33 casos-índice (23 do sexo masculino e 10 do sexo feminino), com idade entre 3 anos e 37 anos, selecionados aleatoriamente do Ambulatório de Otorrinolaringologia, com deficiência auditiva sensorioneural não-sindrômica. Destes casos-índice, foram avaliados 8 parentes (4 do sexo masculino e 4 do sexo feminino), com idade entre 11 anos e 45 anos, que também apresentam deficiência auditiva sensorioneural não-sindrômica. Portanto, foram avaliadas 33 famílias, com pelo menos um membro com deficiência auditiva, constituindo um total de 41 indivíduos deficientes, sendo o projeto aprovado pelo Comitê de Ética, Protocolo nº 4429/2000. Cada paciente foi submetido à completa anamnese para investigar idade de início da deficiência auditiva, presença de outros casos na família e excluir a possibilidade de causas ambientais: infecções materno-fetais, complicações perinatais, meningites, uso de drogas ototóxicas, trauma acústico. Os exames físicos, otorrinolaringológico e sistêmico e exames complementares foram realizados para se excluir sinais sugestivos de formas sindrômicas de deficiência auditiva (especialmente dismorfismo crânio-facial, alterações tegumentares, anomalias de origem branquial, cardíaca, tireoidianas, distúrbios da visão, etc.). Além disso, os pacientes foram submetidos à avaliação oftalmológica (incluindo fundoscopia), testes vestibulares e tomografia computadorizada de osso temporal. Portanto, toda a avaliação clínica foi realizada para se excluir pacientes com deficiência auditiva causada por fatores ambientais, malformações congênitas de orelha interna ou por síndromes genéticas. Os pacientes foram audiologicamente testados por audiometria de tom puro, realizada no Ambulatório de Fonoaudiologia e incluídos aqueles com deficiência auditiva sensorioneural não-sindrômica classificada como leve (25-40 dB), moderada (41-60 dB), grave (61-80 dB) ou profunda (>81 dB)18.
A análise molecular foi realizada no Centro de Biologia Molecular, após extração de DNA a partir de sangue total, a qual foi realizada na Unidade de Pesquisa em Genética e Biologia Molecular, com o kit de extração de DNA genômico (GFXTM Genomic Blood DNA Purification Kit, Amersham Pharmacia Biotech Inc.), conforme protocolo do fabricante. Para a detecção da mutação 35delG foi realizada a técnica da reação em cadeia da polimerase alelo-específico - AS-PCR ("Allele-Specific PCR"), utilizando-se iniciadores ou "primers" específicos para a técnica8. Também foram sintetizados "primers" denominados controles A (direto) e B (inverso), para co-amplificação do gene GJB2 com um segmento do gene amelogenina homólogo ao cromossomo X-Y sendo utilizados, portanto, como controles internos de amplificação19. Além disso, foi realizada a técnica da PCR para detecção da mutação Delta(GJB6 - D13S1830), com "primers" específicos20, nas amostras heterozigotas e nas que não apresentaram a mutação 35delG. Os produtos da AS-PCR e da PCR foram analisados por eletroforese em gel de agarose 1,5% em tampão TBE 1X, contendo brometo de etídio, na concentração de 0,5mg/mL, submetidos à iluminação ultravioleta, para confirmar o sucesso da reação e o gel, fotodocumentado. As amostras que não apresentaram as mutações estudadas, em ambos os alelos, ou as heterozigotas, foram submetidas ao seqüenciamento automático.
Foram sintetizados dois pares de "primers"21, obtendo-se a amplificação do gene GJB2 em dois fragmentos, para a reação da PCR anterior ao seqüenciamento. Os fragmentos amplificados foram purificados utilizando-se o Kit Wizard SV Gel and PCR Clean-UP System (PROMEGA) e as reações de seqüenciamento foram corridas, em ambas as direções, no seqüenciador automático ABI PRISMTM 377 (Perkin Elmer) sendo utilizado o BigDyeTM Terminator Cycle Sequencing Kit V2.0 Ready Reaction (ABI PRISM/PE Biosystems). As seqüências obtidas, ao término do seqüenciamento automático, foram analisadas e comparadas com a seqüência normal com o auxílio do programa Gene Runner V3.05, para alinhamento das seqüências dos nucleotídeos, e do programa Chromas V1.45, para a edição dos eletroferogramas.
Para a revisão dos dados moleculares e audiométricos dos pacientes, o presente estudo, de caráter retrospectivo, portanto, foi submetido e, posteriormente, aprovado pelo Comitê de Ética, Protocolo nº 2813/2004.
Foram calculadas porcentagens com os desvios padrões das mesmas, sendo os resultados expressos em %(DP%).
RESULTADOS
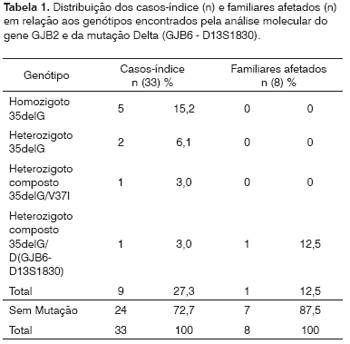
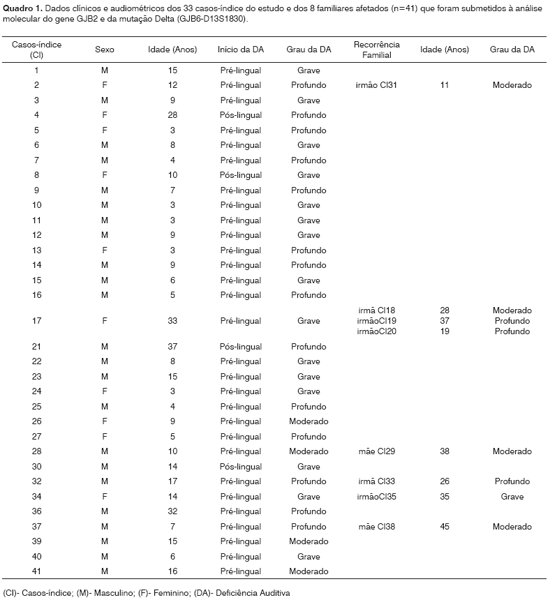
A Tabela 1 apresenta os resultados gerais dos genótipos obtidos após análise molecular realizada nos 33 casos-índice e nos 8 familiares afetados. Os dados clínicos e audiométricos obtidos dos casos-índice tais como sexo, idade, época de início e grau de perda da deficiência auditiva e recorrência familial, são descritos no Quadro 1.
Foram encontradas, portanto, as prevalências de 27,3% (DP%=7,7) da mutação 35delG nos casos-índice analisados (9/33), de 21,2% (DP%=5,03) de alelos (14/66) com a mutação 35delG e de 12,5% (DP%=11,69) nos familiares afetados. Para cada uma das mutações, V37I e Delta(GJB6-D13S1830), foi encontrada a prevalência de 3% (DP%=2,96).
Pelas audiometrias realizadas nos pacientes com deficiência auditiva (n=41), encontrou-se os seguintes resultados em relação aos graus de perda: grau profundo - 17 pacientes (41,5%, DP%=7,69); grau grave - 16 pacientes (39,0%, DP%=7,61); grau moderado - 8 pacientes (19,5%, DP%=6,18). Dos 5 pacientes homozigotos para 35delG, um apresentou grau profundo (2,43%, DP%=2,40), três apresentaram grau grave (7,31%, DP%=4,06) e um paciente apresentou grau moderado (2,43%, DP%=2,40). Dos dois pacientes heterozigotos para 35delG, um (2,43%, DP%=2,40) apresentou grau grave e o outro (2,43%, DP%=2,40) apresentou grau moderado. O paciente heterozigoto composto 35delG/V37I (2,43%, DP%=2,40) e os dois heterozigotos compostos 35delG/Delta (GJB6-D13S1830) apresentaram grau grave (4,87%, DP%=3,36). O predomínio das freqüências audiométricas foi de 4000 a 8000 Hertz (Hz). Todos os pacientes com a mutação 35delG apresentaram deficiência auditiva de início pré-lingual.
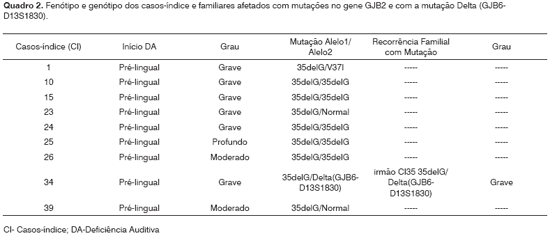
A correlação fenótipo/genótipo dos casos-índice e familiares afetados, com a mutação diagnosticada, está representada no Quadro 2.
DISCUSSÃO
No presente estudo, foram encontradas prevalências da mutação 35delG de 27,3% nos casos-índice analisados, de 21,2% de alelos com a mutação e de 12,5% nos familiares afetados. Foi determinada, também, prevalência de 3% para cada uma das mutações, V37I e Delta (GJB6-D13S1830), encontradas no estudo. Estes resultados são concordantes com estudos já realizados e descritos na literatura, em várias populações22-26. A relativa contribuição da mutação 35delG para a deficiência auditiva não-sindrômica, nessas populações, variou de 0% (Oman, Koréia, Japão) a 70% (Itália, Espanha, Grécia), demonstrando a heterogeneidade genética existente entre os diversos países, apesar de alguns desses estudos terem sido baseados em pequeno número de pacientes, além dos critérios de investigação da deficiência e os métodos de rastreamento da mutação terem sido diferentes entre os mesmos23,27-30.
Estudos recentes encontraram uma deleção de 342 mil pares de bases (342 Kb), próxima ao gene GJB6 [D(GJB6 - D13S1830)], sugerindo que essa mutação pode causar deficiência auditiva não-sindrômica recessiva ou por uma deleção homozigota ou por uma penetrância digênica da deleção, no gene GJB6, associada a uma mutação no gene GJB2 em trans, nos casos heterozigotos24. A maioria dos casos de deficiência auditiva genética resulta de mutações em um único gene, mas cada vez mais, um maior número de casos está sendo identificado como tendo dois genes envolvidos31. Um estudo multicêntrico, realizado em nove países, demonstrou que a mutação Delta (GJB6-D13S1830) é mais freqüente na França, Espanha, Israel, Reino Unido incluindo o Brasil, variando de 5,9% a 9,7% de todos os alelos estudados de pacientes com DFNB1, estando presente em cerca de 50% dos pacientes heterozigotos, na Espanha32. No presente estudo, foi encontrada a prevalência de 3,0% da deleção no gene GJB6 em heterozigose com a mutação 35delG, sendo estes dados concordantes com os da literatura24,32.
No Brasil, foram encontradas as prevalências de 2,24% (1:44,6) de heterozigotos para a mutação 35delG, em estudo realizado em neonatos, na região de São José do Rio Preto, SP33 e a prevalência de 0,97% de heterozigotos (1:103), em um rastreamento, também em neonatos, realizado na região de Campinas, SP34. Em outro estudo realizado, mas em pacientes com deficiência auditiva, mutações no gene GJB2 foram encontradas em 33,5% dos casos sendo que, somente a mutação 35delG foi identificada em 84,2% dos alelos mutantes14. A metodologia utilizada no presente estudo, AS-PCR e seqüenciamento automático, foi semelhante às três referências, anteriormente citadas, mas foi encontrada uma variação na freqüência dos alelos com a mutação 35delG. Este fato pode ser explicado pela diferença na amostra ou talvez pela composição étnica da população brasileira ser altamente heterogênea, ocorrendo a miscigenação entre vários grupos étnicos, principalmente, caucasóides e africanos, podendo ocorrer diferenças na prevalência, em diferentes regiões do país35.
De acordo com a literatura, as análises do gene GJB2 em pacientes com deficiência auditiva freqüentemente demonstram heterozigoze em cerca de 10% a 42% dos casos, a despeito da maioria das mutações ter caráter recessivo23,36,37. No presente estudo, encontrou-se freqüência de 12,1% de casos-índice heterozigotos, resultado concordante com os da literatura23,36,37.
De acordo com os resultados audiométricos, no presente estudo foram encontrados 41,5% dos pacientes com deficiência auditiva de grau profundo, 39,0% dos pacientes de grau grave e 19,5% com deficiência de grau moderado, com predomínio nas altas freqüências (4000 - 8000 Hz), e os pacientes homozigotos e heterozigotos para 35delG, predominando nos graus moderado-grave, sendo este padrão concordante com os dados da literatura17,38,39. A variabilidade nos graus da deficiência auditiva em pacientes homozigotos para 35delG é grande. A maioria das deficiências autossômicas recessivas tem bastante consistência fenotípica, mesmo entre irmãos, o que não é observado para o gene GJB2-35del, principalmente, nos casos heterozigotos. Isso sugere a possibilidade de outros fatores modularem a expressão do gene mutante40. Uma intrigante possibilidade é que pode haver um segundo gene conexina que compartilha funcionalmente com o gene GJB2. É concebível que uma segunda proteína conexina possa atuar como um substituto sob certas condições. Talvez existam genes modificadores em outros locais ou influências ambientais que ativam ou inativam as regiões promotoras do gene. Como a proteína Cx26 está envolvida na homeostase iônica da orelha interna, alguns destes pacientes podem ser capazes de ouvir, por apresentarem uma perda auditiva moderada, sugerindo mecanismos homeostáticos alternativos ou compensatórios. Alterações na proteína Cx26 podem afetar, adversamente, o desenvolvimento do sistema auditivo, resultando em variações no grau de perda ou assimetria38. Influências ambientais, tais como ruídos, drogas ototóxicas, podem ser aditivos ou sinergistas com os defeitos causados por mutações no gene GJB2, aumentando assim, a perda auditiva17,38,41.
As avaliações clínicas dos pacientes deste estudo não sugerem que causas ambientais sejam os maiores fatores, uma vez que se identificou um padrão de transmissão autossômico recessivo nos casos com a mutação 35delG. Além disso, os achados audiométricos destes nove pacientes, por terem apresentado audiometria, de caráter não-progressiva, semelhante às encontradas nos pacientes referidos na literatura23,42-44, com diagnóstico de deficiência auditiva causada por mutação no gene GJB2, confirmam que o gene está envolvido na etiologia da deficiência auditiva dos pacientes deste estudo com expressão fenotípica semelhante à da literatura23,42-44. Em aproximadamente um terço dos casos de deficiência auditiva por mutações no gene GJB2, encontra-se padrão audiométrico de caráter progressivo de perda auditiva, contrariando os dois terços dos casos com o típico padrão não-progressivo. Isso significa que uma criança com perda moderada pode evoluir para profunda e as terapias podem ser diferentes entre esses dois tipos de graus de perda. As famílias que têm uma criança com perda moderada-grave ou profunda poderão se beneficiar pelas análises do gene GJB238.
A mutação 35delG é fácil para se detectar e o teste é viável. Entretanto, uma vez que grande parte dos pacientes com a mutação 35delG são homozigotos, análises mais abrangentes do gene GJB2 serão necessárias, em uma grande proporção dos casos, para distinguir os portadores heterozigotos comuns (portadores sãos) daqueles pacientes heterozigotos que apresentam a DFNB1. Essas análises e pesquisas mais abrangentes [incluindo as análises de toda a região codificante, a região promotora, a região não-codificante do gene e análises da mutação Delta (GJB6-D13S1830)], conforme realizado no presente estudo, devem ser direcionadas pelas características clínicas da DFNB143,45.
De acordo com a literatura, os testes moleculares associados aos dados audiométricos podem predizer que um significante número de pacientes, com a mutação 35delG, terá deficiência de grau moderado-grave e outros são esperados terem grau profundo29,44,46, conforme encontrado neste estudo. Portanto, apesar da casuística pequena, o padrão audiométrico foi concordante com o da literatura, o que possibilitou a correlação genótipo-fenótipo nos dez pacientes da amostra (9 casos-índice e 1 familiar afetado), isto é, os pacientes com mutação 35delG apresentaram perdas moderadas-graves a profundas e não progressiva. Mas é necessário um estudo multicêntrico para se verificar a real expressão fenotípica na população brasileira relacionada à mutação 35delG. O conhecimento do genótipo possibilitará aos médicos, fonoaudiólogos, educadores, auxiliados por médicos-geneticistas, aconselharem os pais, mais apropriadamente, a fim de avaliar o risco de uma futura criança poder ter deficiência auditiva semelhante. As crianças diagnosticadas antes dos seis meses de idade e com um bem-sucedido tratamento com amplificação terão chances muito maiores de desenvolver, normalmente, a fala e a linguagem.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Morton NE. Genetic epidemiology of hearing impairment. Ann N Y Acad Sci 1991;630:16-31.
2. Mustafa T, Arnos KS, Pandya A. Advances in hereditary deafness. Lancet 2001;358:1082-90.
3. Skvorak Giersch AB, Morton CC. Genetic causes of nonsyndromic hearing loss. Curr Opin Pediatr 1999;11(6):551-7.
4. Petit C. Genes responsible for human hereditary deafness: symphony of a thousand. Nature Genet 1996;14:385-91.
5. Van Camp G, Willems PJ, Smith RJH. Nonsyndromic hearing impairment: unparalleled heterogeneity. Am J Hum Genet 1997;60:758-64.
6. Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang, JN, Parry G, Mueller, RF, Leigh IM. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997;387:80-3.
7. Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, Kimberling WJ. Novel mutations in the connexin 26 gene (GJB2) that cause autossomal recessive (DFNB1) hearing loss. Am J Hum Genet 1998;62:792-9.
8. Scott DA, Kraft ML, Carmi R, Ramesh A, Elbedour K, Yari Y, Srisailapathy CRS, et al. Identification of mutation on the connexin 26 gene that cause autossomal recessive nonsyndromic hearing loss. Hum Mutat 1998;11:387-94.
9. Gabriel H, Kupsch P, Sudendey Jr, Winterhager E, Jahnke K, et al. Mutations in the connexin 26/GJB2 gene are the most common event in non-syndromic hearing loss among the German population. Hum Mutat 2001;17:521-2.
10. Van Camp G, Smith RJH. Na Hereditary Hearing Loss Homepage [Site na Internet]. Disponível em: http://webhost.ua.ac.be/hhh/. Acessado em 2006.
11. Zelante L, Gasparini P, Estivill X, Melchionda S, D'Agruma L, Govea N, Mila M, Della Monica M, et al. Connexin 26 mutations associated with the most common form of non-syndromic neurosensory autossomal recessive deafness (DFNB1) in Mediterraneans. Hum Molec Genet 1997;6:1605-9.
12. Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, D'Agruma L, Mansfield E, Rappaport E, et al. Connexin 26 mutations in sporadic and inherited sensorineural deafness. Lancet 1998;351:394-8.
13. Antoniadi T, Gronskov K, Sand A, Pampanos A, Brondum-Nielsen K, Petersen MB. Mutation analysis of the GJB2 (connexin 26) gene by DGGE in greek patients with sensorineural deafness. Hum Mutat 2000;16:7-12.
14. Oliveira CA, Maciel-Guerra AT, Sartorato EL. Deafness resulting from mutations in the GJB2 (connexin 26) gene on Brazilian patients. Clin Genet 2002;61:354-8.
15. Kammen-Jolly K, Ichiki H, Scholtz AW, Gsenger M, Kreczy A, Schrott-Fischer A. Connexin 26 in human fetal development of the inner ear. Hear Res 2001;160(1-2):15-21.
16. Denoyelle F, Marlin S, Weil D, Moatti L, Chauvin P, Garabedian EN, Petit C. Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin 26 gene defect: implications for genetic counselling. Lancet 1999;17(9161):1298-303.
17. Cryns K, Orzan E, Murgia A, Huygen PLM, Moreno F, del Castilo I, et al. A genotype-phenotype correlation for GJB2 (connexin 26) deafness). J Med Genet 2004;41:147-54.
18. World Health Organization. Report of the informal working group on prevention of deafness and hearing impairment programme planning. Geneva: WHO, 1991. 22p
19. Antoniadi T, Gronskov K, Sand A, Pampanos A, Brondum-Nielsen K, Petersen MB. Mutation analysis of the GJB2 (connexin 26) gene by DGGE in Greek patients with sensorineural deafness. Hum Mutat 2000;16:7-12.
20. del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, et al. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 2002;346:243-9.
21. Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, Kimberling WJ. Novel mutations in the connexin 26 gene (GJB2) that cause autossomal recessive (DFNB1) hearing loss. Am J Hum Genet 1998;62:792-9.
22. Sobe T, Vreugde S, Shahin H, Berlin M, Davis N, et al. The prevalence and expression of inherited connexin 26 mutations associated with non-syndromic hearing loss in the Israeli population. Hum Genet 2000;106:50-7.
23. Wilcox SA, Saunders K, Osborn AH, Arnold A, Wunderlich J, et al. High frequency hearing loss correlated with mutations in the GJB2 gene. Hum Genet 2000;106:399-405.
24. del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, et al. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 2002;346:243-9.
25. Frei K, Szuhai K, Lucas T, Weipoltshammer K, Schofer C, Ramsebner R, et al. Connexin 26 mutations in cases of sensorineural deafness in eastern Austria. Eur J Hum Genet 2002;10:427-32.
26. Pampanos A, Economides J, Iliadou V, Neou P, Leotsakos P, Voyiatzis, et al. Prevalence of GJB2 mutations in prelingual deafness in the Greek population. Int J Pediatr Otorhinolaryngol 2002;65:101-8.
27. Gasparini P, Estivill X, Volpini V, Totaro A, Castellvi-Bel S, et al. Linkage of DFNB1 to non-syndromic neurosensory autosomal-recessive deafness in Mediterranean families. Eur J Hum Genet 1997;5:83-8.
28. Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, D'Agruma L, Mansfield E, Rappaport E, et al. Connexin 26 mutations in sporadic and inherited sensorineural deafness. Lancet 1998;351:394-8.
29. Kenna MA, Wu B-L, Cotanche DA, Korf BR, Rehm HL. Connexin 26 studies in patientes with sensorineural hearing loss. Arch Otolaryngol Head Neck Surg 2001;127:1037-42.
30. Simsek M, Al-Wardy N, Al-Khayat A, Shanmugakonar M, Al-Bulushi T, Al-Khabory M, et al. Absence of deafness associated connexin 26 (GJB2) gene mutations in the Omani population. Hum Mutat 2001;18:545-6.
31. Nance WE. The genetics of deafness. Ment Retard Disabil Res Rev 2003;9:109-19.
32. del Castillo I, Moreno-Pelayo MA, del Castillo FJ, Brownstein Z, Marlin S, Adina Q, et al. Prevalence and Evolutionary Origins of the del(GJB6-D13S1830) Mutation in the DFNB1 Locus in Hearing Impaired Subjects: a Multicenter Study. Am J Hum Genet 2003;73:1452-8.
33. Piatto VB, Oliveira CA, Alexandrino F, Pimpinati CJ, Sartorato EL. Perspectivas para triagem auditiva genética: rastreamento da mutação 35delG em neonatos. J Pediatr 2005;81:139-42.
34. Sartorato EL, Gottardi E, Oliveira CA, Magna LA, Annichio-Bizzacchi JM, Seixas CA, Maciel-Guerra AT. Determination of the frequency of the 35delG in Brazilian neonates. Clin Genet 2000;58:339-40.
35. Oliveira CA, Alexandrino F, Abe-Sandes K, Silva Jr WA, Maciel-Guerra AT, Magna LA, Sartorato EL. Frequency of 35delG in the GJB2 gene in samples of Caucasians, Asians and African Brazilians. Hum Biol 2004;76:313-6.
36. Pandya A, Arnos KS, Xia XJ, Welch KO, Blanton SH, Friedman TB, et al. Frequency and distribution of GJB2 (connexin 26) and GJB6 (connexin 30) mutations in a large North American repository of deaf probands. Genet Med 2003;5:295-303.
37. Stevenson VA, Ito M, Milunsky JM. Connexin-30 deletion analysis in connexin-26 heterozygotes. Genet Test 2003;7:151-4.
38. Cohn ES, Kelley PM, Fowler TW, Gorga MP, Lefkowitz, et al. Clinical studies of families with hearing loss attributable to mutations in the connexin 26 gene (GJB2/DFNB1). Pediatrics 1999;103:546-50.
39. Murgia A, Orzan E, Polli R, Martella M, Vinazi C, Leonardi E, Arslan E, Zacchello F. Cx26 deafness: mutation analysis and clinical variability. J Med Genet 1999;36:829-32.
40. Marlin S, Garabedian E-N, Roger G, Moatti L, Matha N, Lewin P, Petit C, Denoyelle F. Connexin 26 gene mutations in congenitally deaf children. Arch Otolaryngol Head Neck Surg 2001;127:927-33.
41. Rabionet R, Zelante L, Lopez-Bigas N, DAgruma L, Melchionda S, Restagno G, et al. Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum Genet 2000;106:40-4.
42. Cohn ES, Kelley PM. Clinical phenotype and mutations in connexin 26 (DFNB1/GJB2), the most commom cause of childhood hearing loss. Am J Med Genet 1999;89:130-6.
43. Denoyelle F, Marlin S, Weil D, Moatti L, Chauvin P, Garabedian EN, Petit C. Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin 26 gene defect: implications for genetic counselling. Lancet 1999;17:1298-303.
44. Engel-Yeger B, Zaaroura S, Zlotogora J, Shalev S, Hujeirat Y, Carrasquilo M, Barges S, Pratt H. The effects of a connexin 26 mutation - 35delG - an oto-acoustic emissions and brainstem evoked potentials: homozygotes and carriers. Hear Res 2002;163:93-100.
45. Mustapha M, Salem N, Delague V, Chouery E, Ghassibeh M, Rai M, Loiselet J, Petit C, Megarbane A. Autosomal recessive non-syndromic hearing loss in the Libanese population: prevalence of the 30delG mutation and report of two novel mutations in the connexin 26 (GJB2) gene. J Med Genet 2001;38:e36.
46. Yoshinaga-Itano C, Sedey AL, Coulter DK, Mehl AL. Language of early-and later-identified children with hearing loss. Pediatrics 1998;102:1161-71.
1 Doutor, Professor Adjunto II-D do Departamento de Otorrinolaringologia e Cirurgia de Cabeça e Pescoço, FAMERP.
2 Graduando 5º Ano Medicina, FAMERP.
3 Mestre, Chefe do Serviço de Fonoaudiologia do Departamento de Otorrinolaringologia e Cirurgia de Cabeça e Pescoço, FAMERP.
4 Livre-docente, Chefe do Departamento de Otorrinolaringologia e Cirurgia de Cabeça e Pescoço, FAMERP.
5 Mestre, Sub-Chefe Departamento de Otorrinolaringologia e Cirurgia de Cabeça e Pescoço, FAMERP.
6 Doutor, Chefe do Centro de Biologia Molecular e Engenharia Genética, CBMEG-UNICAMP.
Faculdade de Medicina de São José do Rio Preto, SP - FAMERP.
Endereço para correspondência: Vânia Belintani Piatto - Rua Santina Figliagi Ceccato 450 ap 23-A. Vila Itália São José do Rio Preto SP 15.035-180.
BIC-FAMERP (Bolsa de Iniciação Científica - FAMERP).
Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) da RBORL em 31 de agosto de 2006. cod. 3367.
Artigo aceito em 2 de novembro de 2006.
Imprimir: ![]()