

Ano: 2005 Vol. 71 Ed. 1 - Janeiro - Fevereiro - (20º)
Seção: Relato de Caso
Páginas: 107 a 110
 PDF PT
PDF PT Síndrome de Treacher Collins com atresia coanal: relato de caso e revisão de suas características
Treacher Collins syndrome with choanal atresia: a case report and a review of its characteristics
Autor(es): Eduardo C. Andrade1 , Vanier S. Júnior1, Ana L. S. Didoni1, Priscila Z. Freitas1, Araken F. Carneiro2 , Fabiana R. Yoshimoto3
Palavras-chave: síndrome de Treacher Collins, atresia coanal, obstrução vias aéreas.
Keywords: Treacher Collins syndrome, choanal atresia, airway obstruction.
Resumo:
A Síndrome de Treacher Collins ou disostose mandibulofacial apresenta-se com deformidades crânio-faciais, tendo expressão e severidade variável. É uma malformação congênita que envolve o primeiro e segundo arcos branquiais. A Síndrome de Treacher Collins é rara e sua incidência está estimada em uma faixa de 1:40000 a 1:70000 nascidos vivos. Esta síndrome é caracterizada por anormalidades dos pavilhões auriculares, hipoplasia dos ossos da face, obliqüidade antimongolóide das fendas palpebrais com coloboma palpebral inferior e fissura palatina. A Síndrome de Treacher Collins raramente está associada com atresia coanal. Estes pacientes são apropriadamente acompanhados por uma equipe multidisciplinar que inclui cirurgiões crânio-faciais, oftalmologistas, fonoaudiologistas, cirurgiões dentistas e otorrinolaringologistas. Relatamos neste artigo um caso raro de Síndrome de Treacher Collins com atresia coanal, uma revisão da patologia e intervenção multidisciplinar.
Abstract:
Treacher Collins Syndrome - or mandibulofacial dysostosis - is a rare condition that presents several craniofacial deformities of different levels. This is a congenital malformation involving the first and second branchial arches. Incidence is estimated to range between 1-40,000 to 1-70,000 of live births. The disorder is characterized by abnormalities of the auricular pinna, hypoplasia of facial bones, antimongoloid slanting palpebral fissures with coloboma of the lower eyelids and cleft palate. Treacher Collins Syndrome is rarely associated with choanal atresia. A multidisciplinary team, including craniofacial surgeon, ophthalmologist, speech therapist, dental surgeon and otorhinolaryngologist, is the most appropriate setting to manage these patients. This study reports a rare case of Treacher Collins Syndrome with choanal atresia, presenting literature review and multidisciplinary intervention.
![]()
INTRODUÇÃO
Embora Thomson tenha sido o primeiro a referenciar esta síndrome em 1846, foi E. Treacher Collins quem descreveu seus componentes essenciais em 1900. Franceschetti e Klein, em 1949, realizaram intensivos estudos da síndrome. Além de Síndrome de Treacher Collins, epônimo preferido pela literatura inglesa, é referida também como disostose mandibulofacial, Síndrome de Berry e Síndrome de Franceschetti-Zwahlen-Klein1-4.
A incidência estimada da Síndrome de Treacher Collins varia de 1:40000 a 1:70000 nascidos vivos1,5. Não existe predileção entre os sexos e as raças. Tem transmissão autossômica dominante de expressividade variável. O gene para a Síndrome de Treacher Collins foi mapeado na porção distal do braço longo do cromossomo 5 (5q31.3-q33.3). Sua expressão fenotípica provavelmente resulta de uma malformação congênita envolvendo o primeiro e segundo arcos branquiais, bilateralmente6-8.
A síndrome pode apresentar grande variação de diferentes formas clínicas. Obliqüidade antimongolóide das fendas palpebrais, hipoplasia malar, hipoplasia mandibular, malformações dos pavilhões auriculares, coloboma palpebral inferior, surdez condutiva e fissura palatina estão entre as manifestações clínicas mais características1,4. Os traços clínicos são usualmente simétricos e bilaterais2. Atresia de coanas ocasionalmente é encontrada na Síndrome de Treacher Collins9.
Anomalias craniofaciais predispõem à obstrução de vias aéreas e síndrome da apnéia obstrutiva do sono. Estes pacientes devem receber acompanhamento de equipe multidisciplinar que inclui cirurgiões craniofaciais, fonoaudiólogos, cirurgiões-dentistas e otorrinolaringologistas, visando um apropriado controle das vias aéreas10. Relatamos um caso de Síndrome de Treacher Collins com atresia coanal bilateral e revisão da patologia.
RELATO DO CASO
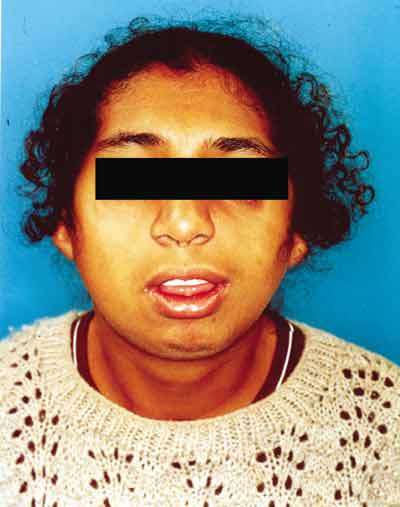
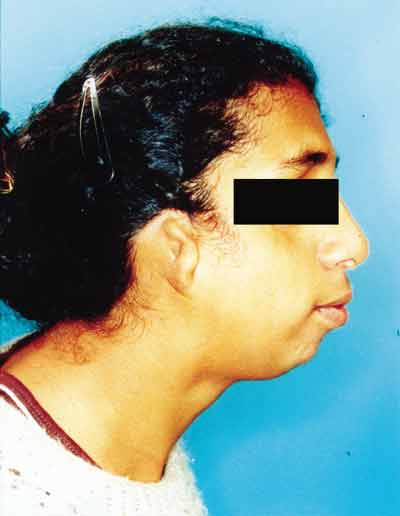
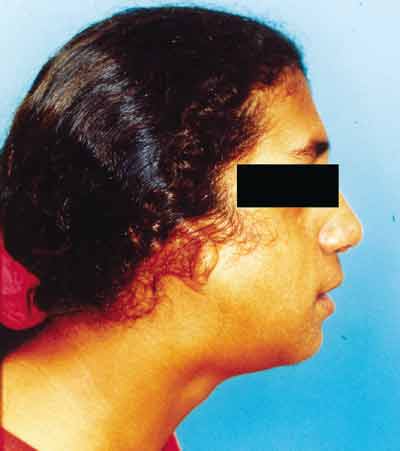
Paciente feminino, 18 anos, procurou nosso serviço de otorrinolaringologia com queixa de obstrução nasal e respiração bucal. Apresentava ao exame obliqüidade antimongolóide das fendas palpebrais, hipoplasia de arco zigomático e mandibular, coloboma palpebral inferior bilateral e malformação dos pavilhões auriculares (Figuras 1 e 2). Apresenta déficit de linguagem e aproveitamento escolar regular. Desvio de septo nasal anterior e posterior foram observados pela rinoscopia.
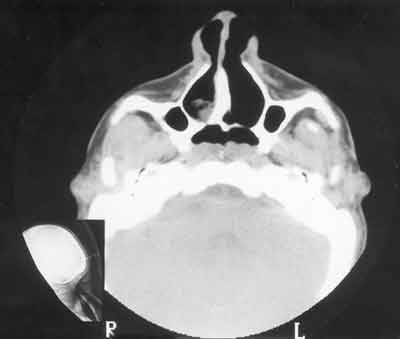
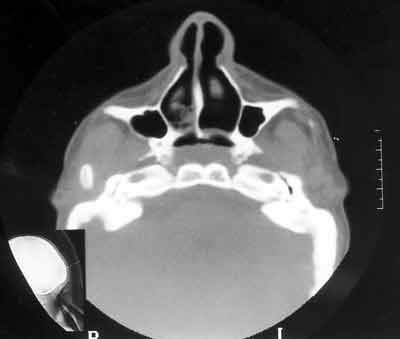
A tomografia computadorizada de rinofaringe demonstrou obstrução de coanas bilateral por lâmina constituída por tecido ósseo e membranoso, sendo que a lâmina direita predomina o tecido ósseo e a esquerda membrana de partes moles (Figuras 3 e 4). A nasofibroscopia confirmou o diagnóstico de atresia coanal. A paciente foi submetida à septoplastia e correção de atresia coanal pelo acesso transpalatal, sendo colocado um dreno torácico em cada cavidade nasal, visando manter a permeabilidade das coanas. Paciente evoluiu com processo inflamatório na região do vestíbulo nasal direito e deiscência da sutura em palato. Foi feita a remoção dos drenos e nova sutura do palato. Apresentou no pós-operatório episódios de apnéia noturna, mesmo mantendo coanas pérvias. Foi submetida à cirurgia de mentoplastia de avanço pela equipe da cirurgia ortognática do serviço e uvulopalatoplastia pela equipe de otorrinolaringologia (Figura 5). Apresentou boa evolução com melhora do padrão respiratório. Está em acompanhamento sem alterações respiratórias.
Figura 1. Sinais clínicos da Síndrome de Treacher Collins.
Figura 2. Sinais clínicos da Síndrome de Treacher Collins (perfil).
Figura 3. CT mostrando atresia coanal (predomínio de tecido ósseo à direita) - corte axial.
Figura 4. CT mostrando atresia coanal (predomínio de tecido membranoso à esquerda) - corte axial.
Figura 5. Resultado após mentoplastia de avanço.
DISCUSSÃO
A Síndrome de Treacher Collins é uma deformidade rara, ocorrendo em aproximadamente 1:50000 nascidos vivos2,6. Apresenta grande variedade de alterações, sendo que as manifestações clínicas mais freqüentes são: obliqüidade antimongolóide das fendas palpebrais (89%), hipoplasia malar (81%), hipoplasia mandibular (78%), malformação dos pavilhões auriculares (77%) e coloboma palpebral inferior (69%)4. Já a associação de atresia coanal e a síndrome ocorre de maneira ocasional, sendo manifestação pouco freqüente na Síndrome de Treacher Collins9.
Algumas entidades clínicas como a Síndrome de Godenhar (Displasia Oculo-Auriculo-Vertebral), Disostose Acrofacial de Nager e Síndrome de Miller são diagnósticos diferenciais da Síndrome de Treacher Collins2,4. O acometimento simétrico e bilateral é uma característica importante da Síndrome de Treacher Collins11.
Pacientes com anomalias craniofaciais estão predispostas à obstrução de vias aéreas. A participação de uma equipe multidisciplinar é fundamental para um correto manejo das vias aéreas desses pacientes. Na Síndrome de Treacher Collins, os pacientes podem apresentar obstrução crônica de vias aéreas superiores, fato este que acarreta prejuízo para a alimentação e o sono, além do aumento do número de hospitalizações. É freqüente o encontro de micrognatia, postura anormal da língua, hipoplasia de faringe e estreitamento de laringe e traquéia levando à obstrução respiratória e síndrome da apnéia obstrutiva do sono6,10,12.
A tomografia computadorizada de rinofaringe é um método diagnóstico muito útil para identificar obstrução anatômica de vias aéreas13. É capaz de demonstrar com grandes detalhes atresia coanal ou outra possível malformação que leve à obstrução. A incidência de atresia coanal é estimada em 1:5000 nascidos vivos, prevalece no sexo feminino (2:1) e metade dos casos estão associadas com outras anormalidades (craniofaciais, cardiovasculares ou abdominais). A atresia é óssea em 90% dos casos e nos 10% remanescentes é membranosa. É mais comum a atresia coanal unilateral. A correção endonasal, transpalatal e trans-septal são os acessos utilizados. Cada um apresenta vantagens e desvantagens, sendo a idade do paciente fator importante para a escolha da técnica. O acesso transpalatal é preferido em crianças mais velhas e adultos14. O procedimento cirúrgico de avanço mandibular é descrito com sucesso para a melhoria da obstrução respiratória em pacientes com retrognatia e estreitamento de vias aéreas superiores10.
COMENTÁRIOS FINAIS
A Síndrome de Treacher Collins é uma rara doença hereditária autossômica dominante que apresenta uma grande variedade de manifestações clínicas. A presença de atresia coanal bilateral em pacientes com a síndrome é raramente observada.
A abordagem atual para as deformidades clínicas na Síndrome de Treacher Collins visa a correção funcional, a correção estética e a necessidade de apoio psicossocial, tendo a participação conjunta de uma equipe multidisciplinar formada por otorrinolaringologistas, cirurgiões craniofaciais, oftalmologistas, fonoaudiólogos, psicólogos e cirurgiões-dentistas para obter tal objetivo. Além das anomalias anatômicas e fisiológicas, associa-se nos pacientes com a Síndrome de Treacher Collins o estigma social pelas severas deformidades faciais.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Marres HAM, Cremers CWRJ, Dixon MJ, Huygen PLM, Joosten FBM. The Treacher Collins syndrome: a clinical, radiological, and genetic linkage study on two pedigrees. Arch Otolaryngol Head Neck Surg 1995; 121(5):509-14.
2. Dixon MJ. Treacher Collins syndrome. J Med Genet 1995; 32(10):806-8.
3. Anil S, Beena VT, Ankathil R, Remani P, Vijayakumar T. Mandibulofacial dysostosis: case report. Aust Dent 1995; 40(1):39-42.
4. Aguiar RS, Santos CS. Síndrome de Treacher Collins. Rev Port Estomatol Cir Maxilofac 1989; 30(2):137-42.
5. Posnick JC. Treacher Collins syndrome: perspectives in evaluation and treatment. J Oral Maxilofac Surg 1997; 55(10):1120-33.
6. Argenta LC, Iacobucci JJ. Treacher Collins syndrome: present concepts of the disorder and their surgical correction. World J Surg 1989; 13(4):401-9.
7. Edery P, Manach Y, Le Merrer M, Till M, Vignal A, Lyonnet S, Munnich A. Apparent genetic homogeneity of the Treacher Collins - Franceschetti syndrome. An J Med Genet 1994; 52(2):174-7.
8. Shah FA, Ramakrishna S, Ingle V, Dada JE, Al Khabori M, Murty PSN. Treacher Collins syndrome with acute airway obstruction. Int J Pediatr Otorhinolaryngol 2000; 54(1):41-3.
9. Moorman-Voestermans K, Vos A. Bilateral choanal atresia in two members of one family. J Pediatr Surg 1983; 18(2):175-6.
10. Perkins JA, Sie KCY, Milczuk H, Richardson MA. Airway management in children with craniofacial anomalies. Cleft Palate Craniofac J 1997; 34(2):135-40.
11. Posnick JC, Ruiz RL. Treacher Collins syndrome: current evaluation, treatment and future directions. Cleft Palate Craniofac J 2000; 37(5):434.
12. Holinger LD, Weiss KS. Diagnosis and management of airway obstruction in craniofacial anomalies. Otolaryngol Clin North Am 1981; 14(4):1005-17.
13. Handler SD. Upper airway obstruction in craniofacial anomalies: diagnosis and management. Birth Defects Orig Artic Ser 1985; 21(2):15-31.
14. Maniglia AJ, Goodwin WJJr. Congenital Choanal Atresia. Otolaryngol Clin North Am 1981; 14(1):167-73.
1 Médicos residentes em Otorrinolaringologia do HRAC-USP-Bauru.
2 Médico e preceptor da residência em Otorrinolaringologia do HRAC-USP-Bauru.
3 Médico otorrinolaringologista.
Instituição: Hospital de Reabilitação de Anomalias Crânio Faciais - USP - Bauru.
Endereço para correspondência: Eduardo Carvalho de Andrade - R: Prof. Gerson Rodrigues, 7-51 ap. 1601 Vila Universitária Bauru SP 17012-310.
Tel (0xx14) 234-5462 - E-mail: eduardocandrade@uol.com.br
Trabalho apresentado no 36º Congresso Brasileiro de Otorrinolaringologia realizado em Florianópolis, SC em Novembro de 2002.
Imprimir: ![]()