

Ano: 1972 Vol. 38 Ed. 1 - Janeiro - Abril - (11º)
Seção: Notícias e Comentários
Páginas: 88 a 95
Aterogênese
Autor(es):
Rubem Rodrigues* e
Cortos Antonio M. Gottschall**
![]()
SINOPSE: Conceituação e patologia da aterosclerose; estudo sobre o ateroma.
1 - Patologia dinâmica: A Aterosclerose é uma complexa patologia que se caracteriza, sobretudo, por uma ruptura do equilíbrio dinâmico entre:
a) - as substâncias lipídicas do sangue;
b) - a estrutura e as propriedades metabólicas da parede vascular;
c) - a hemodinâmica entra-arterial(7).
O desequilíbrio surge sempre que um desses fatores esteja alterado ou, com maior intensidade, quando os três modificam seu comportamento si. A conseqüência é uma deposição de lipídios na camada íntima da parede arterial e na região mais interna da camada média, justa-intimalz (Fig. 1).
2 - Esse desequilíbrio depende, em parte, da elevação das substâncias lipídicas do sangue, principalmente o colesterol e seus ésteres, bem como os triglicerfdios, além de, secundariamente, os fosfolipídios.
Os lipídios sanguíneos, por serem insolúveis na água, deslocam-se na corrente circulatória junto com determinadas proteínas, formando complexos lipoproteicos que lhes conferem poder de solubilização. Dessa forma são transportados às unidades funcionais, onde participam de processos metabólicos intracelulares. Tais complexos lipoproteicos constituem-se em quatro grupos fundamentais, reconhecidos pelas diferentes velocidades migratórias eletroforéticas, em função de sua densidade e do tamanho das partículas: alfa-lipoproteínas, pré-betalipoproteínas, beta-lipoproteínas e quilomicra.
Os complexos lipoproteicos distinguem-se não somente por suas propriedades biofísico-químicas mas, sobretudo, pela concentração em diferentes lipídios. Deles depende o poder aterogênico desses complexos. Como as maiores concentrações em ésteres de colesterol e de triglicerol (triglicerídios) encontram-se nas prébeta e beta-lipoproteínas e nas quilomicras, os referidos complexos são os mais importantes no desencadeamento de formação do ateroma.
Baseado no comportamento eletroforético das lipoproteínas, Fredrickson(10), formulou uma classificação da aterosclerose em cinco grupos de objetiva aplicação clínica, em 1965. Mais tarde, em 1968, Strisower(27), utilizando diferentes critérios e procedimentos, estabeleceu uma classificação em quatro grupos que apresenta, entretanto, surpreendente correlação com a classificação de FREDRICKSON, do ponto de vista clínico.
Esse autor lançou mão do procedimento proposto por Gofman(15) que consiste na separação das macro-partículas lipoproteicos por ultracentrifugação, em suas frações Sf.
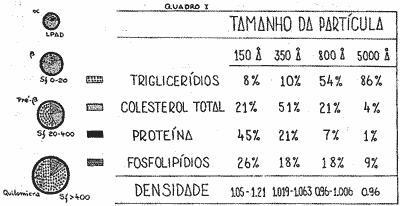
O diagrama e a tabela abaixo mostram a composição das lipoproteínas e seu tamanho relativo (Quadro I).
Esse diagrama permite uma imagem comparativa da eletroforese com a ultracentrifugação. Verifica-se que as porções de menor densidade são mais ricas em triglicerídios (Sf 20-400 e Sf > 400), correspondendo às lipoproteínas pré-beta e quilomicra, de maior tamanho (800 A° a 5000 A°). O colesterol possui sua maior concentração na fração Sf 0-20 que tem tamanho de 350 A°, o qual corresponde às beta-lipoproteínas pela eletroforese.
Fig. 1 - Parede arterialQuadro I
Composição e tamanho das partículas lipoproteicas
Observa-se que os dois métodos se equivalem, pois que embora utilizem diferentes processos, fornecem os mesmos resultados. A desvantagem da ultracentrifugação é o elevado custo do equipamento, compensado pelo baixo custo de manutenção e por possibilitar exames em massa. A eletroforese, que preferentemente deve ser feita em acetato de celulose, tem mais baixo custo de instalação, mas limita a realização de exames em massa.
Ultimamente surgiu um método menos dispendioso que analisa outras propriedades das lipoproteínas para distinguir suas diferentes partículas. Trata-se da mensuração de dispersão da luz refletida através de um nefelômetro, por um simples e rápido cálculo. É um método pouco dispendioso que está ao alcance de qualquer laboratório. Sua eficácia tem sido demonstrada pela similaridade dos resultados em relação aos dois outros métodos.
Pelo que foi exposto, observa-se que, embora seja necessária a formação dos complexos lipoproteicos para o transporte dos lipídios, são fundamentalmente suas porções lipídicas que participam na formação da placa ateromatosa, especificamente o colesterol e os triglicerídios.
As primeiras lesões que aparecem são estrias lineares de coloração amarela, firmemente retidas na substância fundamental da camada íntima da parede arterial. Essas pápulas gordurosas, ou placas, aumentam de volume com a evolução do processo e tornam-se salientes no endotélio arterial. Gradativamente a placa ateromatosa perde sua coloração amarela original, tornando-se mais clara e de aparência cartilaginosa(28).
Com o passar do tempo os ateromas podem apresentar uma ou mais das seguintes complicações:
a) - ulceração
b) - calcificação
c) - trombose
d) - hemorragia
Tais complicações são as responsáveis pela oclusão total da luz arterial, com conseqüente necrose da região que deixa de ser perfundida. Quando esse processo se desenvolve na circulação coronariana, desencadeia o quadro clínico de infarto do miocárdio.
3 - As características da parede arterial desempenham, entretanto, importante função na formação do ateroma. Os estudos de Wissler(32) demonstram que na constituição dessa estrutura existem dois tipos de células, com distintas características, que são individuais para cada pessoa. São as células miointimais e os fibrócitos. As primeiras, que apresentam aspecto de células musculares lisas vacuoladas, por isso também chamadas de células espumosas, possuem ou susceptibilidade ou resistência genética para fagocitarem maior ou menor quantidade de lipídios. São as responsáveis pelo caráter familiar da aterosclerose. Quando sua membrana se rompe, lança os lipídios fagocitados na substância fundamental mucopolisacarídea, dando início à formação da pápula ateromatosa. Essas alterações processam-se em função de, numa primeira instância, decréscimo da capacidade das células miointimais para metabolizar os lipídios; numa segunda fase, proliferação dessas células, agora transformadas em aterócitos que, além de aumentarem de volume, multiplicam-se numericamente. Essas alterações ocorrem francamente nas células geneticamente suscetíveis. Nessas, as lipoproteínas mais pobremente metabolizadas acumulam-se, determinando necrose do aterócito por seu efeito irritante ou tóxico(16).
De sua parte, os fibrócitos, que são histiócitos modificados, são estimulados pelo crescente aumento do derrame lipídioo na substância fundamental, em função da necrose e ruptura do aterócito, tornando-se hiperfuncionantes e desencadeando um processo fibrinogénico, com intensa formação de fibras colágenas(24). Tal reação fibrosa passa a fazer parte do ateroma. Isto é, a partir dessa etapa, junta-se à ateromatose a fibrose ou esclerose, portanto constitui-se a aterosclerose propriamente dita.
Esses conhecimentos tornaram-se viáveis na última década, graças à uma série de investigações elaboradas à luz da ultramicroscopia eletrônica e da histoquímical. Em 1967 já era possível sumarizar a ultraestrutura precoce do ate- roma humano e Chidoni e O'Neal(13) afirmavam que "o principal resultado dos estudos ultraestruturais tinha emergido da célula muscular lisa da substância fundamental (célula miointimal) como a principal e essencial célula tipo no ateroma". Além do reconhecimento dessa célula como dominante na placa aterosclerótica, foi possível identificar grande quantidade de lipídios em seu interior, preponderantemente colesterol e triglicerídios.
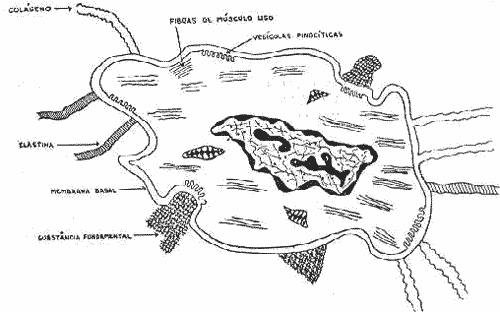
Inúmeras investigações realizadas sobre a célula miointimal tornaram sua estrutura bem conhecida (Fig. 2). É uma célula alongada, com um núcleo central dotado de contornos irregulares(32). Contém miofilamentos em seu citoplasma, distribuídos longitudinalmente. Raras mitocondrias são observadas em torno do núcleo e vesículas pinocitóticas(23) localizam-se, em grande quantidade, junto à membrana citoplasmática. Junto à membrana basilar, ou mesmo partindo dela em direção ao exterior, observam-se fibras colágenas e elastínicas(4).
Esses aspectos ultraestruturais e o achado de lipoproteínas em seu interior(11, 24) em animais que eram alimentados com dieta hiperlipidêmica rica em colesterol permitiram concluir que a essência da aterogênese repousa na célula miointimal ou aterócito. O acúmulo de lipídios, na maioria das vezes, está associado á proliferação e aparente migração celular, seguida de lesão e necrose do aterócito(17).
A presença de mitocôndrias no citoplasma celular e de fibras colágenas e elastínicas em sua membrana basilar permite concluir que elas possuem importantes funções metabólicas. Tal possibilidade foi reforçada por recentes estudos e pesquisas com imunofluorescência direta, que identificaram miosina e actina em: células miointimais de artérias normais e ateroscleróticas humanas(21).
O conceito moderno de aterosclerose fundamenta-se, portanto, numa patologia dinâmica, na qual o aterócito ou célula miointimal desempenha função primordial. A seqüência das alterações que ocorrem na parede arterial seria a seguinte:
- 1) passagem de lipoproteínas, de baixa e ou muito baixa densidade, do sangue para a parede arterial18, em função da propriedade de se combinarem com os mucopolisacarídios da substância fundamenta129, da hiperlipidemia, do aumento da permeabilidade do endotélio3 e ou aumento da pressão hidrostática do sangue (hipertensão arterial, obstáculos localizados, etc.)(8);
- 2) fagocitose das macromoléculas lipoproteínas pelos aterócitos;
- 3) incapacidade do aterócito de metabolizar, total ou parcialmente, alguns ou todos os componentes lipídicos. Parece que os fosfolipídicos seriam parcialmente metabolizados(6), o que não ocorre com o colesterol e os triglicerídios;
- 4) lesão e necrose das células miointimais face à depressão da respiração celular, determinada pela menor difusão de oxigênio dévido ao acúmulo lipídico e por sua ação tóxica. Isso porque a maior parte da glicose consumida pela parede arterial o é por glicólise aeróbica (é insignificante o "efeito Pasteur"), além da fosforilização oxidativa que é de grande importância para a produção de ATP. Observa-se mesmo uma diminuição nas, desidrogenases do ciclo de Krebs, principalmente da desidrogenase succínica(33);
- 5) aumento da síntese de ácidos graxos saturados pela malonil CoA, pela parede arterial, predominantemente palmitato, com conseqüente aumento da esterificação e da síntese de fosfolipídios: esterificação do glicerofosfato, acetilação da lisolecitina e incorporação da fosforil colina da citidina difosfató na lecitina(12, 22, 26).
- 6) proliferação celular por estímulo dos óleos saturados que se esterificam fixamente com o colesterol e o triglicerol(20). Inicia-se um ciclo vicioso que favorece a expansão das alterações ateroscleróticas focais. Aumenta a produção das fibras colágenas pelas células fibrófilas, que se transformam em fibrócitos, marginados por depósitos de ácidos mucopolisacarídios. Segue-se eventual hiperplasia intimal com alterações na substância fundamental;
- 7) necrose celular culminando com acumulação de lipídios cristalizados na substância fundamental (ateroma) que pode evoluir para ulceração, trombose, hemorragia intra-ateromatosa e embolização por desprendimento de fragmento da placa ateromatosa.
4 - As alterações da hemodinâmica intra-arterial são também importantes no desencadeamento do processo aterosclerótico, pois delas depende o aumento do ritmo de perfusão das lipoproteínas de baixa densidade através do endotélio arterial que pode ser determinado por hipertensão arterial(19), aumento da permeabilidade endotelial por anóxia, trauma, toxinas, choque, deposição de fibrinas e agregação plaquetária(8, 9, 14, 30). Essa última parece ter importante função na hiperplasia fibrosa, pois o achado de fibrina na parede arterial aterosclerótica faz supor que as plaquetas seriam o veículo de transporte do fibrinogênio(5).
Não é improvável, também, que as plaquetas, alteradas em sua adesividade endotelial, agregando-se em maior número nas áreas de irritação endotelial, atuem pela sua capacidade de liberar serotinina, histamina e outras aminas vasopressoras que aumentam a permeabilidade endotelial. Há evidências de que as plaquetas alteradas possam penetrar no endotélio e liberar lipídios do seu citoplasmas. Dessa forma, sua ação não estaria restrita somente à sua participação na organização do trombo .mural: contribuiria também para o desenvolvimento do processo aterogênico. Ultimamente, Shimamoto(25) verificou que a bradicinina provoca reação edemato e hiperplásica no endotélio arterial, que seria precursora da aterogênese, favorecendo o depósito lipídico e a formação do ateroma. Ademais, provoca aumento da adesividade e da capacidade aglutinante das plaquetas e leucócitos e inibe a fibrinólise.
5 - Sintetizando as três fases fundamentais que participam na aterogênese, o seguinte esquema pode ser elaborado:
a)
- aumento das células espumosas
- aumento da quantidade das betalipoproteínas de baixa e muito baixa densidade
- alteração de qualidade dos lipídios das lipoproteínas
- diminuição da estabilidade das lipoproteínas
- aumento da tendência coagulante ou da adesividade plaquetária
b)
- depressão do metabolismo das células miointimais (aterófilos ' aterócitos
- proliferação das células miointimais
- acumulação de lipoproteínas mal metabolizadas nas células miontimais ou aterócitos
- lipoproteínas tóxicas ou irritantes nas células miontimais determminando necrose
- estímulo das células fibrófilas, transformadas em fibrócitos, que desencadeiam a hiperplasia fibrosa ou esclerose
c)
- aumento da permeabilidade endotelial devido à hipertensão, aminas vaso-ativas liberadas pelas plaquetas, anóxia, choque, toxinas
- fragmentação ou duplicação da membrana elástica interna
- interrupção da drenagem linfática e dos vasa-vasorum.
SYNOPSIS: Concepts and pathology of atherosclerosis; estadies on human atheroma.
BIBLIOGRAFIA CONSULTADA
1. Altschull, R. Selective studies on arteriosclerosis. Springfield, Charles C. Thomas, 1950.
2. Ashton, W. L. "Definition of atheroma; its pathoiogical sequelae and descriptive epidemiology." In: Human atheroma. London, Willian Heineman, 1967, p. 5.
3. Bragdon, J. H. Chyiomicrons and lipid transport. Annals of New York Academy of Sciences, New York, N. Y., 72:845 et seq., 1959.
4. Buck, R. C. "Histogenesis and morphology of arterial tissue." In: Sanddler, M. & Bonone, G. H. Atherosclerosis and its orïgin. New York, Academic Press, 1963, p. 1.
5. Chandler, A. B. & Hands, R. A. Phagocytized pratelets, a source of lipids in human thrombi and atherosclerotic plaques. Science, New York, N. Y., 134:946 et seq., 1961.
6. Chobanian, A. V. & Hoflander, R. N. Phospholipid synthesis in human arterial intima. Journal of Clinical Investigation, New Haven, Con., 45:932 et seq., 1966.
7. Duff, G. Il. & MacMillan, G: C. Pathology of atherosclerosis. American Journal of Medicine, New York, N. Y., 11 :92 et seq., 1951.
8. Duncan Jr., L. E. et alii. The effect of pressure and stretching on the passage of labeled albumin into canine aortic wall. Journal of Atherosclerosis Research, Amsterdam, 5 : 69 et seq., 1965.
9. Estedy, J. A. & Glagov, S. Altered permeability of the renal artery of the hypertensive rat; an electronamicroscopie study. American Journal of Pathology, Boston, Mass., 43:619 et seq., 1963.
10. Fredrickson, D. S. et alii. Eat transport in lipoproteins; an integrated a aproach to mechanisms on disorders. New England Journal of Medicine, Boston, Mass., 276:34-44; 94-109; 148-56; 215-25; 273-81, 1967.
11. Geer, J. C. et alii. The fine structure of the human atherosclerotic lesions. American Journal of Pathology, Boston, Mass., 31:263 et seq., 1961.
12. Getz, G. S. et alii. The lipid composition and biosynthesis lecithin In atherosclerotic aorta of Rhesus monkey fed three foad fats. Circulation,. New York, N. Y., 36:13 et seq., 1967.
13. Guidoni, J. J. & O'neal, R. M. Recent acivances In molecular pathology; a review of ultrastructure of human atheroma. Journal of Experimental Molecular Pathology, 5:378 et seq., 1967.
14. Glagov, S. et alii. Atherosclerosis of the human aorta and its coronary and renal arteries. Archives of Pathology, Chicago, III., 72:558 et seq., 1961.
15. Gofman, J. W. et alii. Ultracentrifugal studies of lipoproteins of human serum. Journal of Biological Chemistry, Baltimore, Md., 179:973 et seq., 1949.
16. Haust, D. M. et alli. The role of smooth muscle cells in the fibrogenesis of arteriosclerosis. American Journal of Pathology, Boston, Mass., 37:377 et seq., 1960.
17. Haust, D. M. & More, R. H. "Significance of the smooth muscle in atherogenesis." In: fones, R. J. Evaluation of the atherosclerotic plaque. Chicago, University of Chicago Press, 1963. p. 51.
18. Hollander, R. W. et alii. "The metabolism of cholesterol, lipoproteins and acid mucopolysaccharides in normal and atherosclerotic vessels."In: Miras, C. H. et alii. Progress in biochemical pharmacology; recent advances in atherosclerosis. New York, Phiebig, 1968, v.. 4, p. 270.
19. Holman, R. L. et alii. The natural history of atherosclerosis; the aearly aortic lesions as seen in New Orleans in the midle of the 20th Century. American Journal of Pathology, Boston, Mass., 39:209 et seq., 1958.
20. Howard Jr., C. F. De novo svnthesis and elongation of fathy acids by subcellular fractions of monkey aortas. Journal of Lipid Research, New York, N. Y., 9:254 et seq., 1968.
21. Knieren, H. J. et alii. Actomyosin and myosin and the deposition of lipids an serem lipoproteins. Archives of Pathology, Chicago, III, 84:118 et seq., 1967.
22. Portman, O. W. Phospholipid metabolism in subcellular fractions of aortic intimes plus inner media using (I 14 C) fatty acid and (I 14 C) fatty acyt CoA; importante of tysophosphatidylcholine. Circulation, New York, N. Y., 36 :13 et seq., 1967.
23. Rhodin, J. A. G. Fine structure of vascular walls in mammals with special referente to smoth muscle componente. Physiology Review, Baltimore, Md., 42:48 et seq., 1962.
24. Scott, R. E. et alii. Coronary arteries of children and young adults; a comparison of lipids and anatomic features in New Yorkers and East Africans. Journal of Experimental Molecular PathologY, 5:12 et seq., 1966.
25. Shimamoto, T. & Numano, E. Atherogenesis. Tokyo, Excerpta Medica Foundation, s.d.
26. Stein, V. et alii. Enzymic pathway of glycerids and phospholipid synthesis in aortic homogenates. Biochimica et Biophysica Acta, New York, N. Y., 70:33 et seq., 1963.
27. Strisower, E. H. et alii. Treatment of hyperlipidemias. American Journal of Medicine, New York, N. Y., 45:488 et seq., 1968.
28. Thomas, W. A. et alii. "Preproliferative phase" of atherosclerosis in suine fed cholesterol. Archives of Pathology, Chicago, III, 86:621 et seq., 1968.
29. Tracy, R. E. et alii. On the antigenic identy of human serum beta and alpha-2 lipoproteins, and their identification in the aortic intima. Circulation Research, New York, N. Y., 9:472 et seq., 1961.
30. Willis, C. C. Localizing factors in atherosclerosis. Canadian Medical Association Journal, Toronto, 70:1 et seq., 1954.
31. Wissler, R. W. & Wessehnovitch, D. Recent advances in the understanding of the pathogenesis and the reversal of atherosclerosis apud Getz, G. S.; Wesselinovitch, D.; Wissler, R. W. A dynamic pathology of atherosclerosis. American Journal of Medicine, New York, N. Y., 46:655, 1969.
32. Wissler, R. W. The arterial medical cell, smooth muscles or multifunctional mesenchymicme? Journal of Atheroscierosis Research, Amsterdam, 8:201 et seq., 1968.
33. Zempennyi, T. Enzyme biochemistry of the arterial wail. London, Lloid Dulce, 1968.
Retirado da Rev. AMRIGS 15 (4) :293-300 1971.
* Professor Titular da Disciplina de Cardiologia da Faculdade Católica de Medicina de Porto Alegre.
Professor Adjunto do Departamento de Medicina Interna da Faculdade de Medicina da Universidade Federal do Rio Grande do Sul.
Presidente da Fundeação Universitária de Cardiologia, em convênio com o Instituto de Cardiologia do R. G. do Sul.
Membro do "International Council of Epidemiology e Prevention of Cardiovascular Diseases". "
** Coordenador da Unidade de Pesquisa e do Grupo de Estudo em Cardiopatia Isquêmica do Instituto de Cardiologia do Rio Grande do Sul, Fundação Universitária de Cardiologia.
Especialista em Cardiologia pela Sociedade Brasileira de Cardiologia e Associação Médica Brasileira.
Auxiliar de Ensino do Departamento de Medicina Interna da Faculdade de Medicina da Universidade Federal do Rio Grande do Sul.

Imprimir: ![]()
All rights reserved - 1933 /
2026
© - Associação Brasileira de Otorrinolaringologia e Cirurgia Cérvico Facial