

Ano: 2001 Vol. 67 Ed. 4 - Julho - Agosto - (19º)
Seção: Relato de Casos
Páginas: 569 a 572
Aplasia de Michel - Relato de Caso e Revisão de Literatura.
Michel's Aplasia - Case Report and Literature Review.
Autor(es):
Patrícia F. Santos*,
Sebastião Rocha Neto*,
Silvio Caldas Neto**,
Nelson Caldas***,
Alvaro Pontes Júnior****.
Palavras-chave: aplasia de Michel, malformação de orelha interna, surdez congênita
Keywords: Michel's aplasia, inner ear malformation, congenital hearing loss
Resumo:
A aplasia de Michel consiste na ausência total de desenvolvimento da orelha interna. A escassa literatura concernente a essa anomalia traduz sua raridade, tendo sido relatados muito poucos casos clássicos até hoje. O diagnóstico depende de cuidadosa avaliação por imagens. Neste trabalho, apresentamos um caso de aplasia de Michel atendido em nosso Serviço, discorremos sobre as possibilidades etiopatogênicas e alertamos para a necessidade de meios diagnósticos avançados.
Abstract:
Michel's Aplasia consists in total absence of inner ear development. The poor literature concerning this abnormality shows its singularity, since very few classical cases have been related up to now. The diagnosis depends on careful radiological evaluation. In the present paper we reported one case of Michel's Aplasia diagnosed in the ENT clinic of Hospital das Clínicas, UM The authors discussed etiopathogenic possibilities and emphasized the need for advanced diagnostic means.
![]()
INTRODUÇÃO
As malformações da orelha interna afetam aproximadamente 20% dos pacientes com perda auditiva congênita do tipo neurossensorial, sendo que, em cerca de 50% destes, estão envolvidos mecanismos genéticos1.
Subotic e Schuster6 (1978) dividem a surdez neurossensorial congênita em dois grandes grupos: o que reúne alterações no desenvolvimento do labirinto ósseo e membranoso e o que decorre de afecções restritas a estruturas individuais no labirinto membranoso. No primeiro grupo situam-se as malformações congênitas da orelha interna e existem diversas formas propostas para classificá-las. Aqueles autores as subdividem também em dois tipos:
1) Completa ausência da orelha interna (Michel).
2) Anomalias congênitas da cóclea, resultantes de maior ou menor aplasia do modíolo (Mondini).
Ormerod5 divide essas malformações em quatro grupos: Michel, Mondini-Alexander, Bing-Sibeman e Scheibe. Já Schucknecht7 as classifica em Michel, Mondini, Scheibe e Alexander.
Quanto à etiopatogenia, essas malformações podem estar associadas a um defeito do mecanismo de indução, pelo mesoderma cordal e o tubo neural, da diferenciação das estruturas neuroectodérmicas do aparelho auditivo3. Também Steel e colaboradores4 discorrem sobre possíveis alterações no processo de crescimento neuroepitelial na gênese deste tipo de malformação, com base em estudos com indução em animais. Fatores genéticos ainda desconhecidos provavelmente são a chave para o desencadeamento dessas alterações, muito embora sejam descritas anomalias da orelha interna causadas por teratogênicos, como a talidomida.
Dessas anomalias, a mais rara é a de Michel. Esta é uma forma descrita pelo autor de mesmo nome4, em 1863, caracterizada pela ausência completa da orelha interna. Há, entretanto, relatos, na literatura, de casos classificados como aplasia de Michel, que diferem das características relatadas por aquele autor, como constatam Ghazli e colaboradores2, que apresentam dois casos clássicos de aplasia de Michel bilateral em gêmeos.
A repercussão, é óbvio, corresponde à anacusia no ouvido comprometido. Mas a função vestibular não é afetada.
Neste trabalho, descrevemos o que acreditamos ser um quadro clássico de aplasia de Michel, unilateral, em um paciente atendido em nosso Serviço universitário.
APRESENTAÇÃO DE CASO CLÍNICO
I. F. S., do sexo masculino, com 15 anos de idade, apresentou-se no Ambulatório de Otorrinolaringologia do Hospital das Clínicas da Universidade Federal de Pernambuco (HC/ UFPE), com queixa de hipoacusia esquerda desde o nascimento e paralisia facial esquerda instalada aos três anos e seis meses de idade, durante um dos vários episódios de otorréia e otalgia que aconteciam na infância. Negava história de qualquer problema na gestação, parto ou período perinatal. Negava também doenças da infância ou uso de drogas ototóxicas. Não havia tampouco história familiar de surdez.
O exame físico mostrou paralisia facial periférica esquerda com função facial grau V de House-Brackmann. A otoscopia, bem como o restante do exame otorrinolaringológico, foi normal. Também não se detectou nenhum sinal de malformações em outros órgãos ou sistemas. A audiometria mostrou anacusia à esquerda e audição à direita.
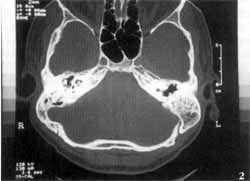
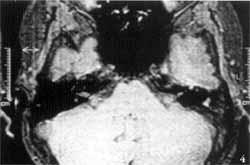
Tomografia computadorizada (TC) dos ossos temporais evidenciou ausência completa das cavidades da orelha interna à esquerda, onde só existia um bloco ósseo correspondente à cápsula ótica. As orelhas média e externa desse lado, bem como o osso temporal direito, eram inteiramente normais (Figuras 1 e 2). Foi realizada ressonância nuclear magnética (RNM), que relevou, no lado afetado, ausência completa do labirinto membranoso, bem como do VIII par craniano. O VIII par pôde ser visto presente (Figuras 3 e 4).
Paciente e família foram então orientados sobre a natureza do problema e as repercussões que ele pode trazer para a vida social e profissional da pessoa, bem como sobre a necessidade de proteção do ouvido normal.
DISCUSSÃO
O estudo das anomalias congênitas da orelha interna torna-se confuso devido ao fato de existirem várias classificações diferentes. É difícil dizer se estas anomalias pertencem a grupos com etiopatogenias distintas ou se não simplesmente formas mais ou menos severas de uma mesma alteração do desenvolvimento embrionário da orelha interna e que apenas receberam nomes diferentes por terem sido descritas por diferentes autores. É bastante provável que esta segunda visão seja a mais verdadeira e que, quanto mais cedo o fato etiopatogênico incidir sobre o desenvolvimento da orelha interna, menos desenvolvida esta estará ao nascimento. Isto já foi bem estudado para a displasia de Mondini, e nada impede que possamos seguir o mesmo tipo de raciocínio para as outras anomalias, incluindo a de Michel. Entretanto, à luz dos novos e revolucionários conhecimentos sobre o código genético humano, é possível que se determinem, em um futuro próximo, genes responsáveis por este ou aquele tipo de evolução da malformação.
O caso descrito por Michel, em 1863, foi revelado em autópsia de uma criança com 11 anos de idade, surda, que havia falecido no Hospital de Strasbourg. A característica mais marcante do caso descrito pelo autor era a ausência não só do labirinto, mas de toda a cápsula ótica, que ficou substituída por fina lâmina óssea que separava a caixa do tímpano da fossa posterior. O nervo facial era preservado, mas o VIII par não existia. As alterações eram iguais nos dois lados.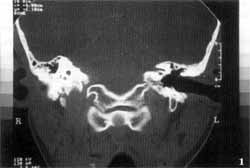
Figuras 1 e 2. Tomografia dos ossos temporais mostrando ausência da orelha à esquerda, com um bloco ósseo correspondendo à cápsula ótica.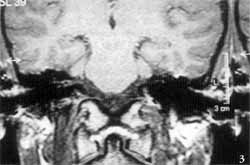
Figuras 3 e 4. Ressonância nuclear magnética dos ossos temporais mostrando ausência completa do labirinto membranoso e do VIII par craniano à esquerda.
Parece que casos idênticos a este descrito por Michel só foram relatados no trabalho de Ghazli e colaboradores7. Estes autores consideram os gêmeos descritos por eles como os únicos casos com a síndrome de Michel clássica e que todos os outros relatos anteriores mostravam ora rudimentos do labirinto membranoso, ora persistência da cápsula ótica, ainda que sem as estruturas membradosas.
O caso por nós descrito enquadra-se nesta última observação, pois é evidente a existência de um bloco de osso compacto separando a orelha média da fossa posterior, e não uma fina lâmina, como uma descrição de Michel.
Entretanto, consideramos esta diferença pouco expressiva em termos de identificação de uma anomalia, mesmo porque achamos que, muito provavelmente, como foi comentado acima, todas essas malformações podem representar uma mesma doença, em graus diferentes de severidade. Na verdade, existe realmente uma diferença mais importante entre a aplasia de Michel e as outras formas, que é, ao nosso ver, a ausência total do labirinto membranoso e do nervo vestibulococlear. Isto pode denunciar uma peculiaridade etiopatogênicana aplasia de Michel, pois significaria, em primeiro lugar, a incidência do fator etiológico numa fase pré-vesícula ótica, ou mesmo pré-placódio ótico; e em segundo lugar, um defeito também nos mecanismos de indução pelo tubo neural, que é, em parte, responsável pela formação do nervo estato-acústico a partir de expansões do notocórdia.
Particularmente em relação à aplasia de Michel, confusão pode ser feita devido a uma avaliação radiológica incompleta. No passado, a falta da TC de alta resolução deve ter induzido a erros diagnósticos, por conta da pobreza das imagens obtidas com os métodos existentes. Rudimentos de orelha interna, por exemplo, poderiam passar despercebidos. Além disso, a TC não permite uma boa avaliação do labirinto membranoso e esta é essencial para a caracterização da aplasia de Michel. Assim, torna-se fundamental o exame através de RNM, que poderá atestar a ausência destas estruturas e do VIII nervo craniano.
No caso apresentado por nós, é evidente a presença de um único nervo na porção superior do conduto auditivo interno rudimentar, que segue o trajeto próprio do nervo facial dentro do osso temporal.
O VIII nervo está claramente ausente.
COMENTÁRIOS FINAIS
Aplasia de Michel é a forma mais grave de malformação da orelha interna, raramente encontrada em sua forma clássica, sobretudo na forma descrita detalhadamente por aquele autor. O caso aqui apresentado traz as principais características desse tipo de malformação, podendo ser denominado como tal. A TC de ossos temporais e a RNM são exames imprescindíveis para o diagnóstico preciso dessa situação.
As malformações da orelha interna ainda oferecem terreno amplo para investigação etiológica. A atual indefinição etiopatogênica dessas anomalias trazem incertezas quanto como classificá-las. Os recentes progressos no mapeamento genético da espécie humana certamente nos trará uma definição mais precisa.
REVISÃO BIBLIOGRÁFICA
1. COSTA, S. S.; CRUZ, O. L. M. - Otologia clínica e cirúrgica. Revinter. Cap. 4. p. 109-111, 2000.
2. GHAZLI, K.; MERITE-DRANCY, A.; MARSOT-DUPUCH, K.; MEYER, B.; JEUNESSE, Y.; CHOUARD, C. H. - À propos de deux cas familiaux de syndrome de Michel. Ann. Otolaryngol. Chir. Cervicofac., 115: 1, 29-34, 1998.
3. MELNIK, A. R.; WEISS, L. - Mesodermal induction defect as a possible cause of ear malfommations. Ann. Otol. Rhinol. Laryngol., 92f 160-4, 1983.
4. MICHEL, E. M. - Memoirs sue less anomalies congenitales de l'oreille interne. Gas. Méd. Strasburg. 3: 55, 1863.
5. ORMEROD, F. C. - The Pathology of congenital deafness. J. Laryngol. Otol., 74: 919-950, 1960.
6. SUBOTIC, R.; SHUSTER, R. - Simultaneous histological, audiological and radiological findings in congenital anomalies of the inner ear. J. Laryngol. Otol., 92 (4): 281-291,1978.
7. SCHUKNECHT, H. F. - Mondini dysplasia: a clinical and pathological study. Ann. Otol. Rhinol. Laryngol. (Suppl 65). 89: 3-23, 1980.
* Médica (o) Residente da Disciplina de Otorrinolaringologia do Hospital das Clínicas da Universidade Federal de Pernambuco - HC/UFPE.
** Professor Adjunto da Disciplina de Otorrinolaringologia do HC/UFPE.
*** Professor Titular da Disciplina de Otorrinolaringologia do HC/UFPE.
**** Estudante de Medicina do HC/UFPE.
Instituição: Universidade Federal de Pernambuco - UFPE - Serviço de Otorrinolaringologia do Hospital das Clínicas da UFPE.
Endereço para correspondência: Patrícia Ferreira dos Santos - Rua Estrada do Encanamento, 702 - Apto 401 - Bloco C - Casa Forte - 52060-210 Recife/ PE.
Telefones: (Residência) (0xx81) 3442-5200 / Celular (0xx81) 9963-9409 - E-mail: pfpimentel@elogica.com.br
Artigo recebido em 23 de janeiro de 2001. Artigo aceito em 6 de fevereiro de 2001.
Imprimir: ![]()